Scanpy分析全流程(含harmonypy整合/细胞周期矫正/双细胞检测及去除)_python去除双细胞
本次走一遍Scanpy全流程,数据集仍为GSE188711,该数据集可至GEO官网下载。
该内容包含:1.10X数据整理和读取;2.Scanpy全流程(过滤降维聚类分群可视化);3.harmonypy整合数据;4.细胞周期矫正;4.scrublet检测双细胞及去除
上一次推文进行了Seurat和h5ad数据相互转化,以及10X多样本数据整理和读取(Python)。推文可见:https://mp.weixin.qq.com/s/JCGnkNKsQhGgfcazj63Mhw
此外,可以向“生信技能树”公众号发送关键词‘单细胞’,直接获取Seurat V5版本的完整代码。
样本整理(10X格式)
1.导入
import osimport shutilimport re2.确定路径
os.getcwd()3.列出目录下所有包含features的文件
fs = [os.path.join(\'GSE188711_RAW\', f) for f in os.listdir(\'GSE188711_RAW\') if \'features\' in f]print(fs)samples1 = fs# [\'GSE188711_RAW/GSM5688707_features_JCA.tsv.gz\', \'GSE188711_RAW/GSM5688706_features_WGC.tsv.gz\', # \'GSE188711_RAW/GSM5688711_features_R_CRC4.tsv.gz\', \'GSE188711_RAW/GSM5688709_features_RS-CRC1.tsv.gz\', # \'GSE188711_RAW/GSM5688710_features_R_CRC3.tsv.gz\', \'GSE188711_RAW/GSM5688708_features_LS-CRC3.tsv.gz\']4.用正则表达式替换字符串,得到样本名
# samples2 = [re.sub(r\'_features\', \'\', f) for f in fs]# samples2 = [re.sub(r\'\\.tsv\\.gz$\', \'\', f) for f in samples2]# print(samples2)samples2 = [re.findall(r\'GSM\\d+\', f)[0] for f in fs]print(samples2)5.建立样本ID到文件路径的映射字典
sample_to_file = {}for filepath in samples1: # 从文件路径里提取样本ID,和samples2格式对应 # 这里用正则匹配 \'GSM\' + 数字,比如 GSM12345 match = re.findall(r\'GSM\\d+\', filepath) if match: sample_id = match[0] sample_to_file[sample_id] = filepathprint(sample_to_file)6.创建outputs文件夹并移动文件
# 创建 outputs 文件夹(如果不存在)os.makedirs(\'outputs\', exist_ok=True)# 遍历 samples2 ,根据样本ID找到对应文件路径进行复制for sample_id in samples2: if sample_id not in sample_to_file: print(f\"警告:找不到样本 {sample_id} 对应的文件!\") continue y = sample_to_file[sample_id] output_dir = os.path.join(\'outputs\', sample_id) os.makedirs(output_dir, exist_ok=True) # 复制并重命名 features 文件 shutil.copy(y, os.path.join(output_dir, \'features.tsv.gz\')) # 对应的 matrix 文件名替换,构建出matrix文件,并转移 matrix_file = y.replace(\'features\', \'matrix\').replace(\'.tsv\', \'.mtx\') shutil.copy(matrix_file, os.path.join(output_dir, \'matrix.mtx.gz\')) # 对应的 barcodes 文件名替换,构建出barcodes文件,并转移 barcodes_file = y.replace(\'features\', \'barcodes\') shutil.copy(barcodes_file, os.path.join(output_dir, \'barcodes.tsv.gz\'))以上就是整理文件的步骤了。
Scanpy全流程
1.导入
import matplotlib.pyplot as pltimport pandas as pdimport scanpy as scimport numpy as npimport anndata as adimport poochimport os设置全局图片风格,dpi100,背景是白色
sc.settings.set_figure_params(dpi=100, facecolor=\"white\")创建文件夹
os.makedirs(\"1-DataPre\",exist_ok=True)2.读取文件
os.getcwd()# \'/Users/zaneflying/Desktop/PythonGSE188711\'设定输入文件的路径
dir_path = \'/Users/zaneflying/Desktop/PythonGSE188711/outputs/\'os.listdir(dir_path)#[\'.DS_Store\',# \'GSM5688711\',# \'GSM5688710\',# \'GSM5688709\',# \'GSM5688707\',# \'GSM5688706\',# \'GSM5688708\']获取样本文件夹名称
# 这里的目的是不读取以.开头的文件samples = [s for s in os.listdir(dir_path) if not s.startswith(\'.\')]samples# [\'GSM5688711\',# \'GSM5688710\',# \'GSM5688709\',# \'GSM5688707\',# \'GSM5688706\',# \'GSM5688708\']正式读取
sce_list = []# 对于具有多个批次的数据集,应单独对每个样品进行质量控制for s in samples : sample_path = os.path.join(dir_path, s) #使用gene_symbols作为ID,使用10x adata = sc.read_10x_mtx(sample_path, var_names=\'gene_symbols\', cache=True) # 过滤:至少5个细胞的基因,至少300个基因的细胞 (对应min.cells和min.features) sc.pp.filter_genes(adata, min_cells=5) sc.pp.filter_cells(adata, min_genes=300) # 给AnnData添加项目名称 adata.obs[\'project\'] = s sce_list.append(adata)# sce_list 是包含多个AnnData对象的列表sce_list# [AnnData object with n_obs × n_vars = 6277 × 18958# obs: \'n_genes\', \'project\'# var: \'gene_ids\', \'feature_types\', \'n_cells\',# AnnData object with n_obs × n_vars = 4398 × 17683# obs: \'n_genes\', \'project\'# var: \'gene_ids\', \'feature_types\', \'n_cells\',# AnnData object with n_obs × n_vars = 6057 × 18601# obs: \'n_genes\', \'project\'# var: \'gene_ids\', \'feature_types\', \'n_cells\',# AnnData object with n_obs × n_vars = 4760 × 19390# obs: \'n_genes\', \'project\'# var: \'gene_ids\', \'feature_types\', \'n_cells\',# AnnData object with n_obs × n_vars = 4431 × 18278# obs: \'n_genes\', \'project\'# var: \'gene_ids\', \'feature_types\', \'n_cells\',# AnnData object with n_obs × n_vars = 6371 × 18581# obs: \'n_genes\', \'project\'# var: \'gene_ids\', \'feature_types\', \'n_cells\']合并文件
adata = sc.concat(sce_list,label=\"sampleid\",index_unique=\'_\')adata.obs_names_make_unique# AnnData object with n_obs × n_vars = 32294 × 16174# obs: \'n_genes\', \'project\', \'sampleid\'保存文件(h5ad)
adata.write(\'./1-DataPre/GSE188711.h5ad\')3.质控
os.chdir(\"./1-DataPre/\")os.getcwd()# \'/Users/zaneflying/Desktop/PythonGSE188711/1-DataPre\'读取文件
adata = sc.read_h5ad(\"GSE188711.h5ad\")给想要的基因打上标签
# mitochondrial genes, \"MT-\" for human, \"Mt-\" for mouseadata.var[\"mt\"] = adata.var_names.str.startswith(\"MT-\")# ribosomal genesadata.var[\"ribo\"] = adata.var_names.str.startswith((\"RPS\", \"RPL\"))# hemoglobin genesadata.var[\"hb\"] = adata.var_names.str.contains(\"^HB[^(P)]\")adata.var # 可以看到每个基因的元数据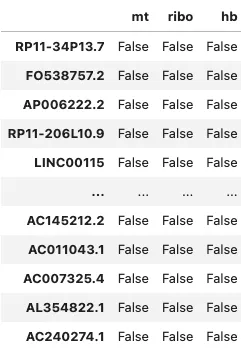
adata.obs # 可以看到每个细胞的元数据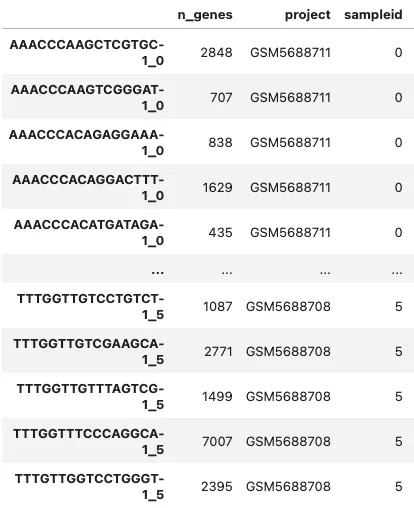
过滤前绘图
# 计算每个细胞针对指定基因集的质量控制指标sc.pp.calculate_qc_metrics( adata, qc_vars=[\"mt\", \"ribo\", \"hb\"], inplace=True, log1p=True)width_cm = 50height_cm = 10plt.figure(figsize=(width_cm / 2.54, height_cm / 2.54))sc.pl.violin( adata, [\"pct_counts_mt\", \"pct_counts_ribo\",\"pct_counts_hb\",\"n_genes_by_counts\", \"total_counts\"], jitter=0.4,# 横向抖动防止重叠 multi_panel=True, # 绘制在单独的子图中 show=False)plt.savefig(\"过滤前.pdf\",bbox_inches=\'tight\') # 不显示就能保存了。。。。show =Falseplt.show()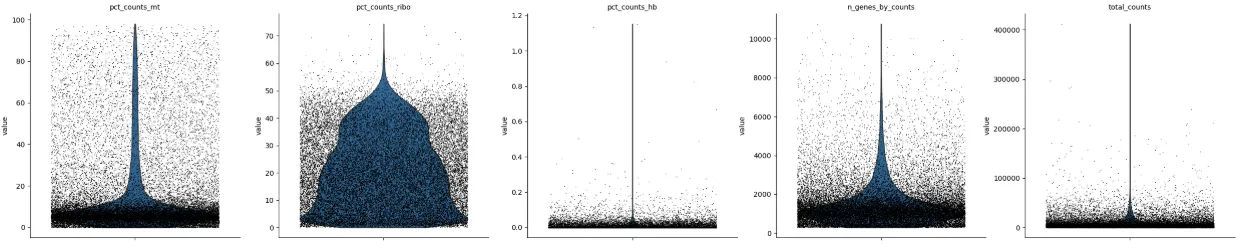
# 对MT、Ribo、Hb、n_cells_by_counts和total_counts数值进行过滤adata = adata[ (adata.obs[\'n_genes_by_counts\'] > 500) & (adata.obs[\'n_genes_by_counts\'] < 6000) & (adata.obs[\'total_counts\'] < 10000) & (adata.obs[\'pct_counts_mt\'] < 10) & (adata.obs[\'pct_counts_ribo\'] < 15) & (adata.obs[\'pct_counts_hb\'] < 1), :]# 这里的:是保留所有的列过滤
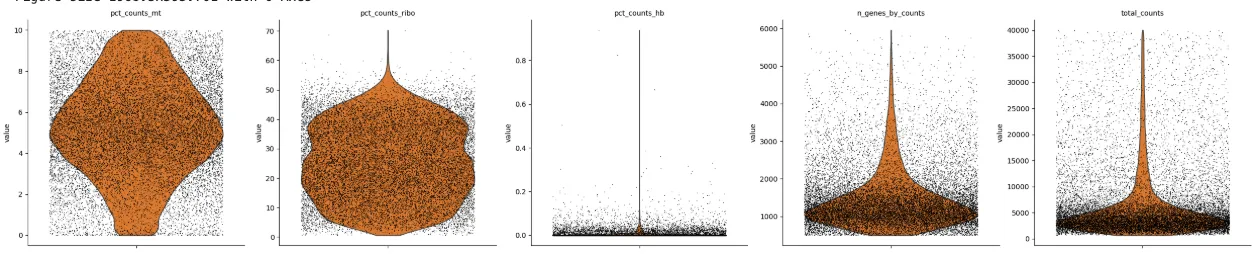
width_cm = 50height_cm = 10plt.figure(figsize=(width_cm / 2.54, height_cm / 2.54))sc.pl.violin( adata, [\"pct_counts_mt\", \"pct_counts_ribo\",\"pct_counts_hb\",\"n_genes_by_counts\", \"total_counts\"], jitter=0.4,# 横向抖动防止重叠 multi_panel=True, # 绘制在单独的子图中 #groupby=\'sampleid\', # 或其他 group 字段 palette=[\"#ff7f0e\"], #[\"#1f77b4\", \"#2ca02c\", \"#d62728\", \"#9467bd\"], show=False)plt.savefig(\"过滤后.pdf\",bbox_inches=\'tight\') # 不显示就能保存了。。。。show =Falseplt.show()颜色自己改
width_cm = 10height_cm = 10plt.figure(figsize=(width_cm / 2.54, height_cm / 2.54))sc.pl.scatter(adata, \"total_counts\", \"n_genes_by_counts\", color=\"pct_counts_mt\",show=False)plt.savefig(\"scatterPlot.pdf\",bbox_inches=\'tight\') plt.show()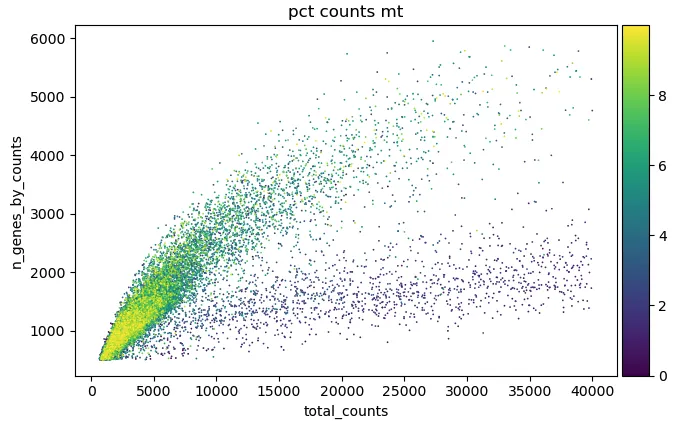
双细胞检测
这里做了之后后面再考虑去不去除
# 使用scrublet进行双细胞检测sc.pp.scrublet(adata, batch_key=\"sampleid\") adata.obs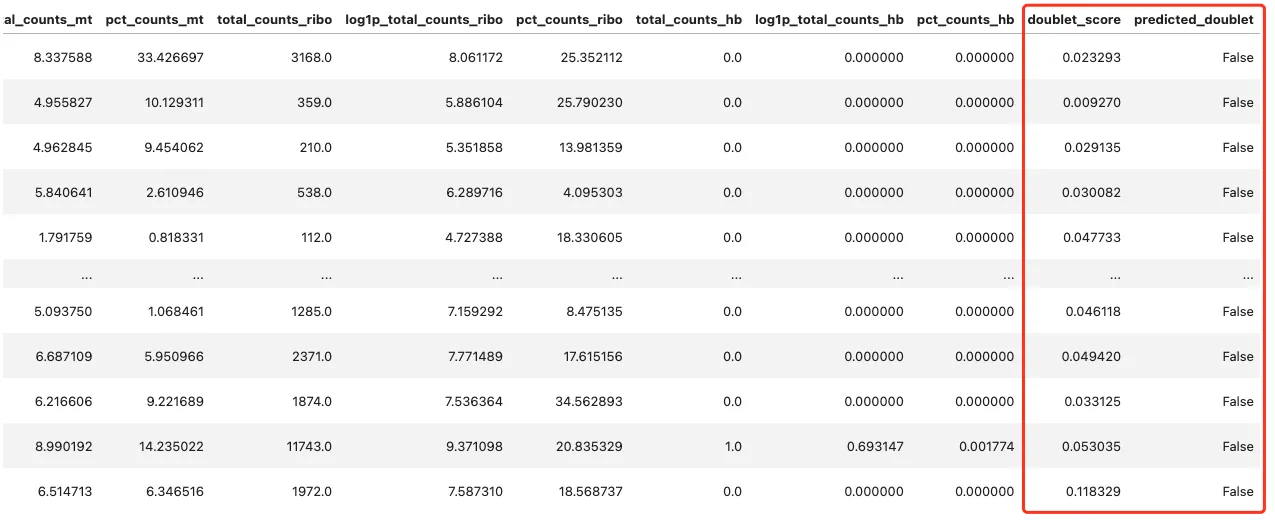
归一化并找高变基因
# 保存原始count矩阵数据 # 这一步也可以不用adata.layers[\"counts\"] = adata.X.copy()adata.layers# Layers with keys: counts# 说明目前layers中已经保存了原始的counts文件,这里就是跟Seurat自动生成不一样归一化
# Normalizing to median total countssc.pp.normalize_total(adata,target_sum=1e4)# Logarithmize the datasc.pp.log1p(adata)把Normallizing和log后的数据赋值到raw中去
# 保存一个原始数据adata.raw = adata.copy()找高变基因
# 全部#sc.pp.highly_variable_genes(adata, n_top_genes=adata.shape[1], batch_key=\"sampleid\")#sc.pl.highly_variable_genes(adata)# 官网推荐sc.pp.highly_variable_genes( adata, min_mean= 0.0125, max_mean= 3, min_disp= 0.5 )sc.pl.highly_variable_genes(adata)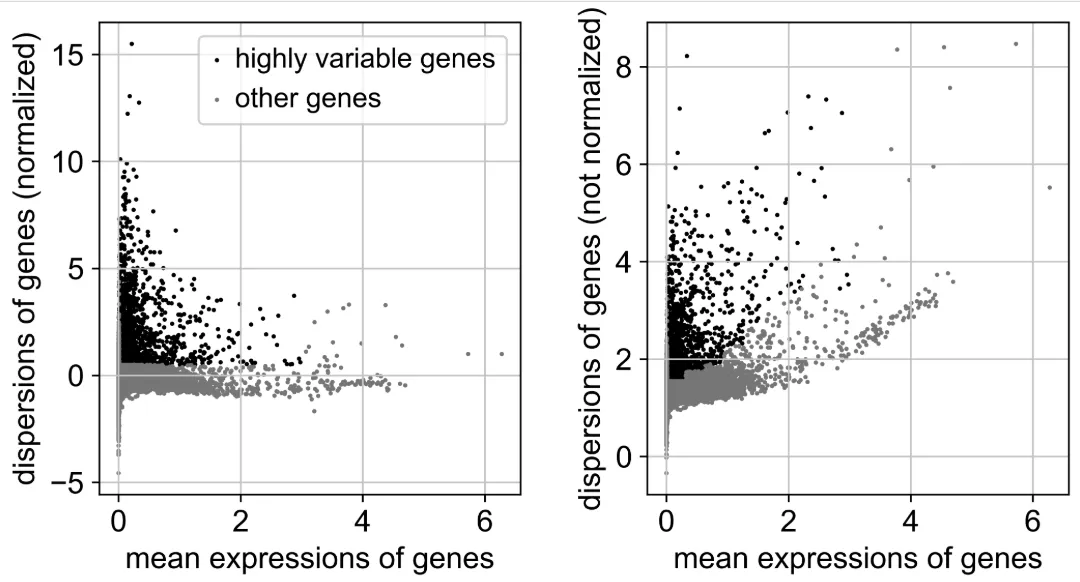
# 保留高变基因,去除其他基因,但其实可以不去除# adata = adata[:, adata.var.highly_variable]# adata细胞周期矫正
# 基因集来自Seurats_genes = [ \"MCM5\", \"PCNA\", \"TYMS\", \"FEN1\", \"MCM2\", \"MCM4\", \"RRM1\", \"UNG\", \"GINS2\", \"MCM6\", \"CDCA7\", \"DTL\", \"PRIM1\", \"UHRF1\", \"HELLS\", \"RFC2\", \"RPA2\", \"NASP\", \"RAD51AP1\", \"GMNN\", \"WDR76\", \"SLBP\", \"CCNE2\", \"UBR7\", \"POLD3\", \"MSH2\", \"ATAD2\", \"RAD51\", \"RRM2\", \"CDC45\", \"CDC6\", \"EXO1\", \"TIPIN\", \"DSCC1\", \"BLM\", \"CASP8AP2\", \"USP1\", \"CLSPN\", \"POLA1\", \"CHAF1B\", \"BRIP1\", \"E2F8\"]g2m_genes = [ \"HMGB2\", \"CDK1\", \"NUSAP1\", \"UBE2C\", \"BIRC5\", \"TPX2\", \"TOP2A\", \"NDC80\", \"CKS2\", \"NUF2\", \"CKS1B\", \"MKI67\", \"TMPO\", \"CENPF\", \"TACC3\", \"SMC4\", \"CCNB2\", \"CKAP2L\", \"CKAP2\", \"AURKB\", \"BUB1\", \"KIF11\", \"ANP32E\", \"TUBB4B\", \"GTSE1\", \"KIF20B\", \"HJURP\", \"CDCA3\", \"CDC20\", \"TTK\", \"CDC25C\", \"KIF2C\", \"RANGAP1\", \"NCAPD2\", \"DLGAP5\", \"CDCA2\", \"CDCA8\", \"ECT2\", \"KIF23\", \"HMMR\", \"AURKA\", \"PSRC1\", \"ANLN\", \"LBR\", \"CKAP5\", \"CENPE\", \"CTCF\", \"NEK2\", \"G2E3\", \"GAS2L3\", \"CBX5\", \"CENPA\"]# 细胞周期矫正sc.tl.score_genes_cell_cycle(adata, s_genes=s_genes, g2m_genes=g2m_genes)# 整合基因cell_cycle_genes = s_genes+g2m_genes矫正前
adata_cc_genes = adata[:, cell_cycle_genes]sc.pp.scale(adata_cc_genes, max_value=10) # 也可以把10设定为nonesc.tl.pca(adata_cc_genes)sc.pl.pca_scatter(adata_cc_genes, color=\'phase\')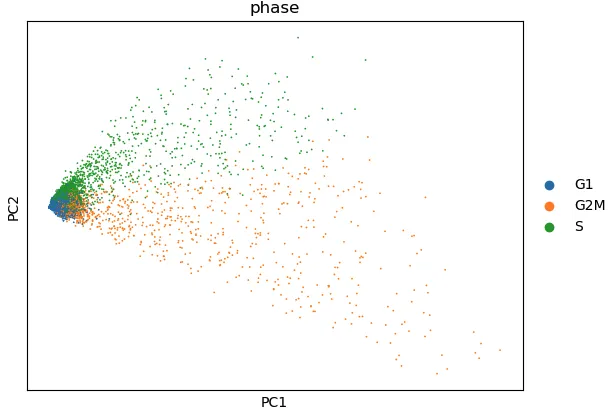
关键的矫正代码
sc.pp.regress_out(adata, [\'S_score\', \'G2M_score\'])矫正后
adata_cc_genes = adata[:, cell_cycle_genes]sc.pp.scale(adata_cc_genes, max_value=10) # 也可以把10设定为nonesc.tl.pca(adata_cc_genes)sc.pl.pca_scatter(adata_cc_genes, color=\'phase\')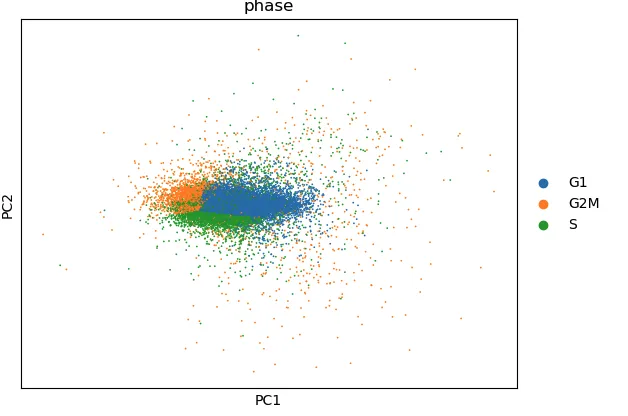
降维聚类分群
# 整体的scale Datasc.pp.scale(adata, max_value=10)# pca降维sc.tl.pca(adata)# 这个图就可以选择合适的PCA数值了sc.pl.pca_variance_ratio(adata,n_pcs=50,log=True)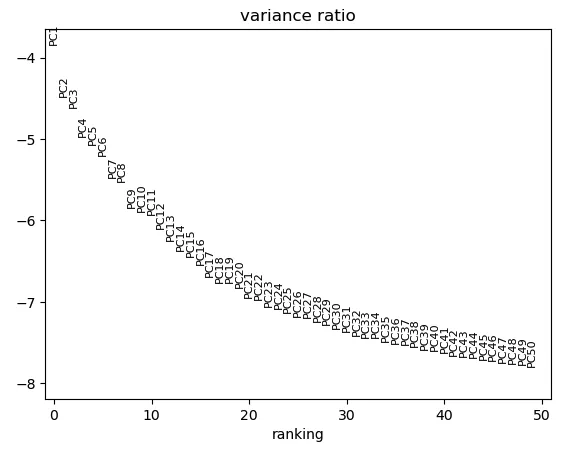
# 按照不同分组检查pca的情况sc.pl.pca( adata, color = [\"project\",\"project\",\"pct_counts_mt\",\"pct_counts_mt\"],# 按照什么参数进行上色 dimensions=[(0,1),(2,3),(0,1),(2,3)], # 按照不同的pca进行展示 ncols =2, size =2,)# 图片不展示构建邻接图
sc.pp.neighbors(adata, n_neighbors=20, n_pcs=15)# umap处理sc.tl.umap(adata)width_cm = 10height_cm = 10plt.figure(figsize=(width_cm / 2.54, height_cm / 2.54))sc.pl.umap( adata, color=\"project\", # Setting a smaller point size to get prevent overlap size=2, show=False)plt.savefig(\"umap_non.pdf\",bbox_inches=\'tight\') # 不显示就能保存了。。。。show =Falseplt.show()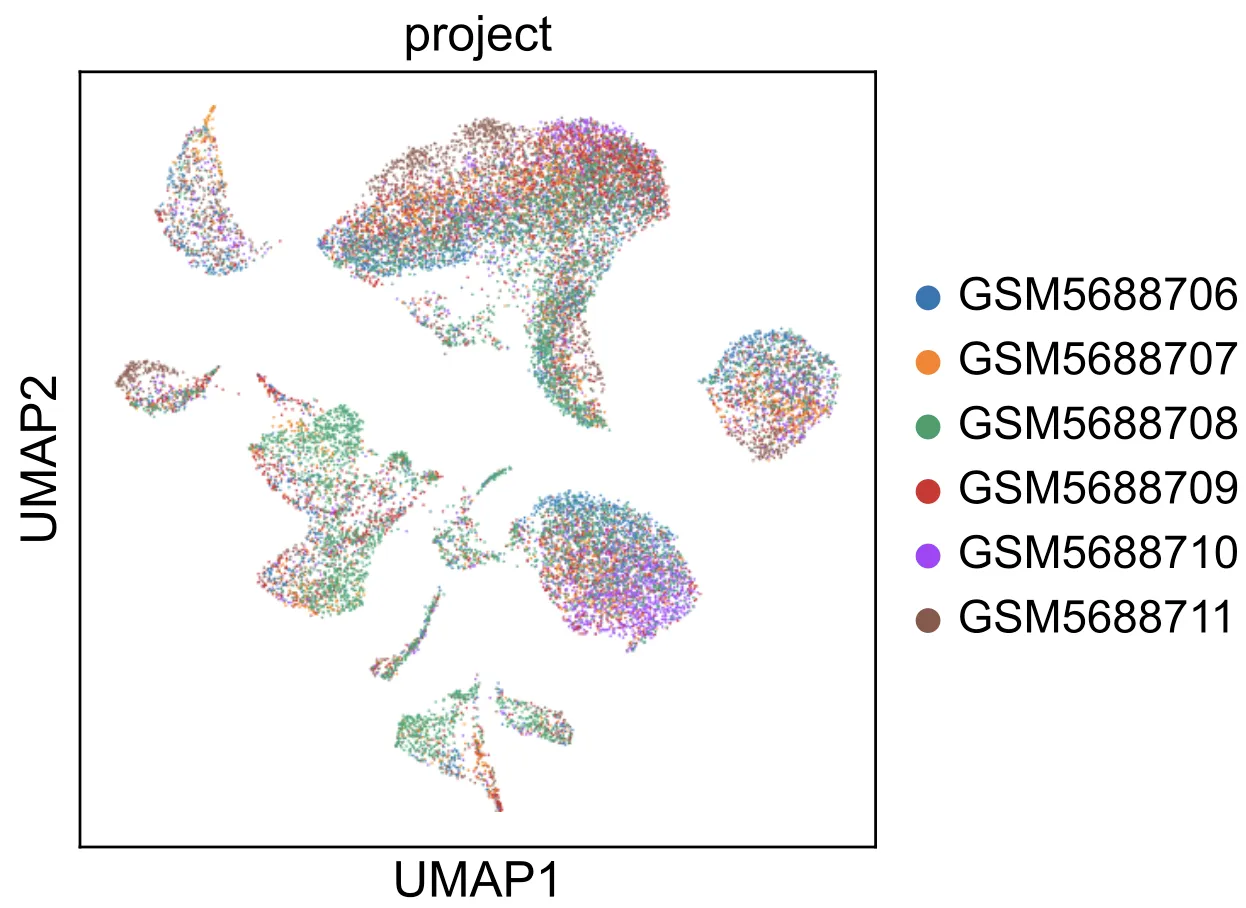
harmony整合样本
import scanpy.external as sce import harmonypy as hm sce.pp.harmony_integrate(adata, \'project\')# Then we calculate a new umap and tsne.sc.pp.neighbors(adata, n_neighbors=20, n_pcs=15, use_rep=\'X_pca_harmony\')sc.tl.umap(adata)# sc.tl.tsne(adata, use_rep=\'X_pca_harmony\')# sc.tl.leiden(adata, resolution=0.5)width_cm = 10height_cm = 10plt.figure(figsize=(width_cm / 2.54, height_cm / 2.54))sc.pl.umap( adata, color=\"project\", # Setting a smaller point size to get prevent overlap size=2, show=False)plt.savefig(\"umap_scanorama.pdf\",bbox_inches=\'tight\') plt.show()
聚类
# 荐莱顿图聚类方法# 生成0.1到2.0的分辨率列表,步长0.1resolutions = np.arange(0.1, 2.1, 0.1)# 对每个分辨率运行Leiden聚类,结果存入adata.obs对应列for res in resolutions: sc.tl.leiden( adata, resolution=res, key_added=f\'leiden_{res:.1f}\', flavor=\'igraph\', n_iterations=2 )# 生成对应的key列表,供绘制聚类树时使用all_keys = [f\'leiden_{res:.1f}\' for res in resolutions]width_cm = 40height_cm = 10plt.figure(figsize=(width_cm / 2.54, height_cm / 2.54))sc.pl.umap(adata, color=[\"leiden_0.2\",\"leiden_0.5\", \"leiden_0.8\",\"leiden_1.0\", \"leiden_1.2\",\"leiden_1.5\",\"leiden_1.8\",\"leiden_2.0\"], show=False)plt.savefig(\"umap_leiden.pdf\",bbox_inches=\'tight\') plt.show()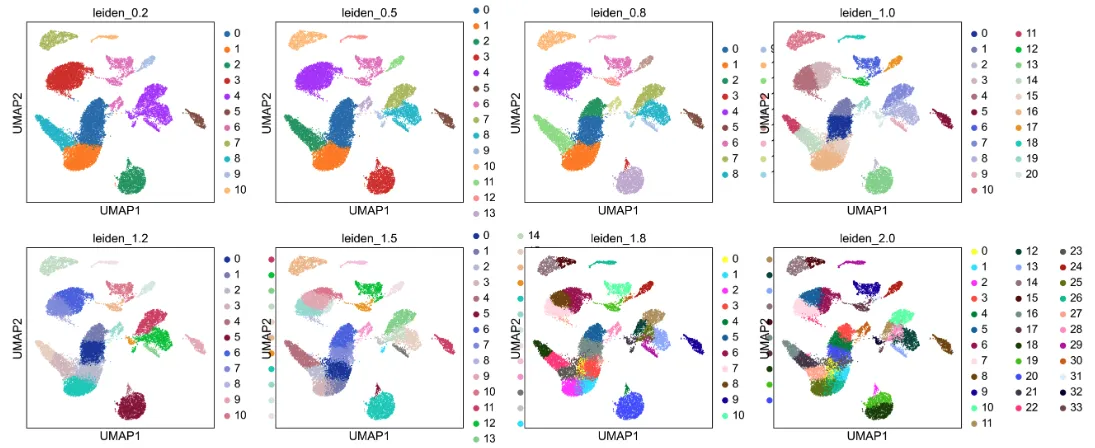
是否需要过滤双细胞
width_cm = 30height_cm = 10plt.figure(figsize=(width_cm / 2.54, height_cm / 2.54))sc.pl.umap( adata, color=[\"leiden\", \"predicted_doublet\", \"doublet_score\"], # increase horizontal space between panels wspace=0.5, size=3, show =False)plt.savefig(\"Doublet_plot.pdf\",bbox_inches=\'tight\')# 不显示就能保存了。。。。show =Falseplt.show()有一点双细胞
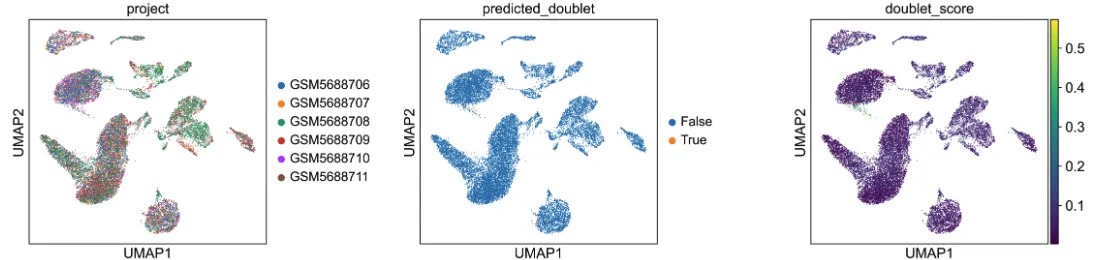
# 过滤双细胞adata = adata[ (adata.obs[\'predicted_doublet\'] == False), :]adata# View of AnnData object with n_obs × n_vars = 19410 × 16174# obs: \'n_genes\', \'project\', \'sampleid\', \'n_genes_by_counts\', \'log1p_n_genes_by_counts\', \'total_counts\', \'log1p_total_counts\', \'pct_counts_in_top_50_genes\', \'pct_counts_in_top_100_genes\', \'pct_counts_in_top_200_genes\', \'pct_counts_in_top_500_genes\', \'total_counts_mt\', \'log1p_total_counts_mt\', \'pct_counts_mt\', \'total_counts_ribo\', \'log1p_total_counts_ribo\', \'pct_counts_ribo\', \'total_counts_hb\', \'log1p_total_counts_hb\', \'pct_counts_hb\', \'doublet_score\', \'predicted_doublet\', \'S_score\', \'G2M_score\', \'phase\', \'leiden_0.1\', \'leiden_0.2\', \'leiden_0.3\', \'leiden_0.4\', \'leiden_0.5\', \'leiden_0.6\', \'leiden_0.7\', \'leiden_0.8\', \'leiden_0.9\', \'leiden_1.0\', \'leiden_1.1\', \'leiden_1.2\', \'leiden_1.3\', \'leiden_1.4\', \'leiden_1.5\', \'leiden_1.6\', \'leiden_1.7\', \'leiden_1.8\', \'leiden_1.9\', \'leiden_2.0\', \'leiden\'# var: \'mt\', \'ribo\', \'hb\', \'n_cells_by_counts\', \'mean_counts\', \'log1p_mean_counts\', \'pct_dropout_by_counts\', \'total_counts\', \'log1p_total_counts\', \'highly_variable\', \'means\', \'dispersions\', \'dispersions_norm\', \'highly_variable_nbatches\', \'highly_variable_intersection\', \'mean\', \'std\'# uns: \'scrublet\', \'log1p\', \'hvg\', \'pca\', \'project_colors\', \'neighbors\', \'umap\', \'leiden_0.1\', \'leiden_0.2\', \'leiden_0.3\', \'leiden_0.4\', \'leiden_0.5\', \'leiden_0.6\', \'leiden_0.7\', \'leiden_0.8\', \'leiden_0.9\', \'leiden_1.0\', \'leiden_1.1\', \'leiden_1.2\', \'leiden_1.3\', \'leiden_1.4\', \'leiden_1.5\', \'leiden_1.6\', \'leiden_1.7\', \'leiden_1.8\', \'leiden_1.9\', \'leiden_2.0\', \'leiden\', \'leiden_colors\', \'leiden_0.2_colors\', \'leiden_0.5_colors\', \'leiden_1.0_colors\', \'leiden_1.2_colors\', \'leiden_0.1_colors\', \'leiden_2.0_colors\', \'leiden_0.8_colors\', \'leiden_1.5_colors\', \'leiden_1.8_colors\', \'predicted_doublet_colors\'# obsm: \'X_pca\', \'X_umap\', \'Scanorama\'# varm: \'PCs\'# layers: \'counts\'# obsp: \'distances\', \'connectivities\'映射基因标志物
marker_genes = { \"Malignant\": [\"KRT14\",\"KRT8\",\"EPCAM\"], # \"KRT5\", \"B/Plasma\": [\"CD79A\", \"MS4A1\", \"TNFRSF13C\", \"TNFRSF17\"], \"T/NK\": [\"CD3D\", \"CD3E\", \"CD4\", \"CD8A\", \"GZMB\", \"NKG7\"], \"Mono/Macro\": [\"CD68\", \"C1QA\", \"C1QB\", \"C1QC\", \"CD14\", \"FCGR3A\", \"FCN1\", \"SPI1\"], \"Pericytes\": [\"RGS5\", \"MYH11\"], \"Mast\": [\"TPSB2\", \"KIT\"], \"Dendritic\": [\"CD1C\", \"FCER1A\", \"LILRA4\"], # \"CLEC9A\" \"Fibroblasts\": [\"LUM\", \"DCN\",\"COL1A1\"], \"Endothelial\": [\"RAMP2\", \"CD34\"], \"VSMCs\":[\"ACTA2\",\"MYH11\"] }width_cm = 30height_cm = 10plt.figure(figsize=(width_cm / 2.54, height_cm / 2.54))sc.pl.dotplot(adata, marker_genes, groupby=\"leiden_1.0\", standard_scale=\"var\", show =False )plt.savefig(\"leiden_1.0_marker.pdf\",bbox_inches=\'tight\')plt.show()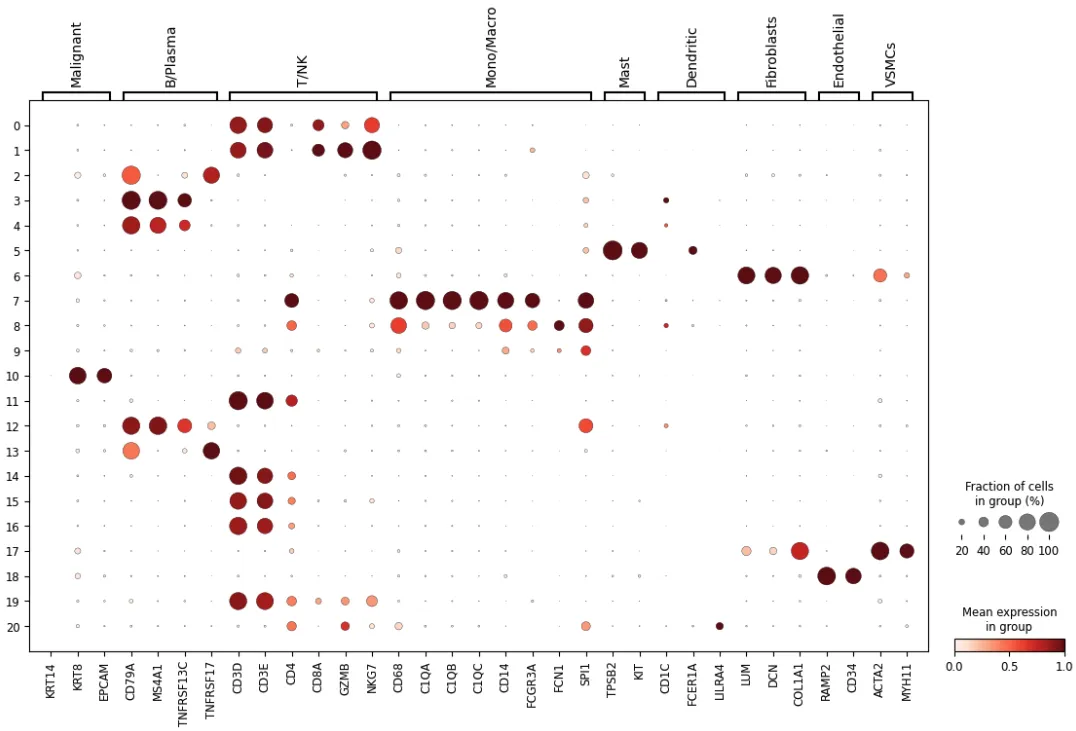
获取集群特异性差异表达基因
sc.tl.rank_genes_groups(adata, groupby=\"leiden_1.0\", method=\"wilcoxon\")width_cm = 30height_cm = 10plt.figure(figsize=(width_cm / 2.54, height_cm / 2.54))sc.pl.rank_genes_groups_dotplot( adata, groupby=\"leiden_1.0\", standard_scale=\"var\", n_genes=5, show =False)plt.savefig(\"leiden_1.0_DEGs.pdf\",bbox_inches=\'tight\')plt.show()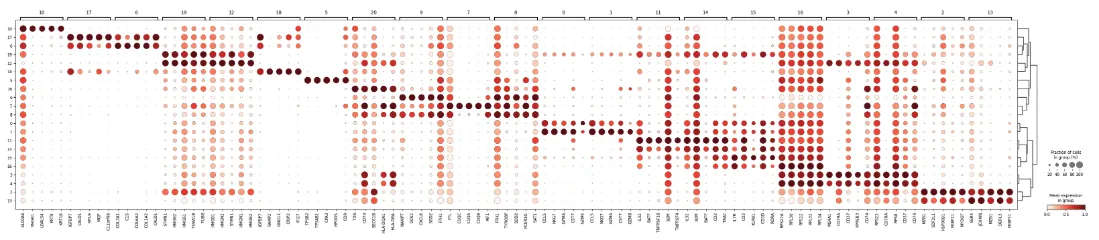
# 取出所有组别的名字,比如 \'0\', \'1\', \'2\' 等groups = adata.uns[\'rank_genes_groups\'][\'names\'].dtype.namesall_dfs = []for group in groups: # 用scanpy自带的函数导出每个组的差异基因结果为DataFrame df = sc.get.rank_genes_groups_df(adata, group=group) df[\'group\'] = group # 加一列表示是哪个组 all_dfs.append(df)# 合并所有组的DataFrameall_results = pd.concat(all_dfs,axis=0) # 0是行合并,1是列合并# 保存为Excel文件all_results.to_csv(\'rank_genes_groups_results.csv\', index=False)sc.get.rank_genes_groups_df(adata, group=\"12\").head(15)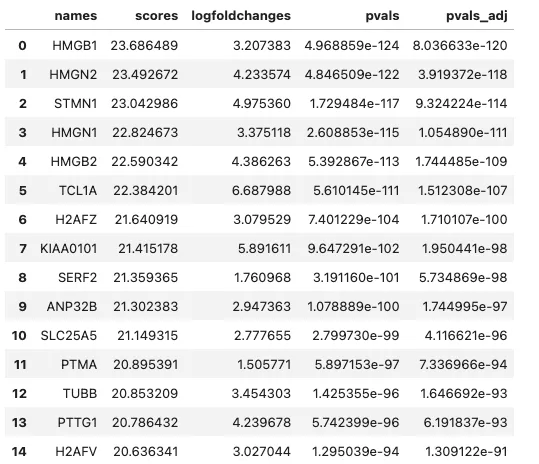
dc_cluster_genes = sc.get.rank_genes_groups_df(adata, group=\"12\").head(5)[\"names\"]sc.pl.umap( adata, color=[*dc_cluster_genes, \"leiden_1.0\"], legend_loc=\"on data\", frameon=False, ncols=3,)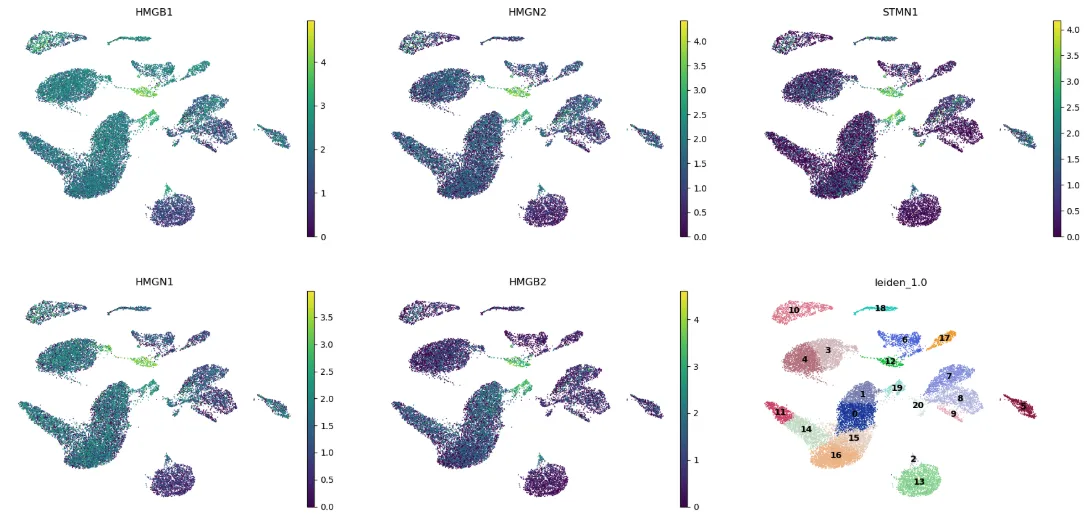
细胞群映射
# 获取所有cluster ID,排序并转成字符串列表(方便后续匹配)cluster_ids = sorted(adata.obs[\'leiden_1.0\'].unique().tolist())cluster_ids = [str(c) for c in cluster_ids] # 转成字符串# 初始化 DataFrame,所有celltype先设为\'Unknown\'celltype = pd.DataFrame({ \'ClusterID\': cluster_ids, \'celltype\': [\'Unknown\'] * len(cluster_ids)})celltypeadata.obs[\'leiden_1.0\'].value_counts()# leiden_1.0# 16 2594# 4 1897# 0 1775# 13 1702# 3 1566# 15 1479# 14 1193# 8 1104# 1 1039# 7 1019# 10 809# 6 698# 11 606# 5 477# 17 290# 18 258# 19 231# 12 222# 20 200# 9 179# 2 72# Name: count, dtype: int64# 根据cluster ID分组赋值细胞类型(字符串类型匹配)# .loc[行条件, 列名] = 新值 语法表示:对满足“行条件”的那些行,在指定列名的位置赋新值celltype.loc[celltype[\'ClusterID\'].isin([\'10\']), \'celltype\'] = \'Malignant cells\'celltype.loc[celltype[\'ClusterID\'].isin([\'3\',\"4\",\"12\"]), \'celltype\'] = \'B cells\'celltype.loc[celltype[\'ClusterID\'].isin([\"2\",\"13\"]), \'celltype\'] = \'Plasma\'celltype.loc[celltype[\'ClusterID\'].isin([\'0\',\'1\',\"11\",\"14\",\"15\",\"16\",\"19\"]), \'celltype\'] = \'T/NK cells\'celltype.loc[celltype[\'ClusterID\'].isin([\'7\',\'8\',\'9\',\"20\"]), \'celltype\'] = \'Mono/Macro cells\'celltype.loc[celltype[\'ClusterID\'].isin([\'6\']), \'celltype\'] = \'Fibroblast\'celltype.loc[celltype[\'ClusterID\'].isin([\'5\']), \'celltype\'] = \'Mast cells\'celltype.loc[celltype[\'ClusterID\'].isin([\'18\']), \'celltype\'] = \'Eendritic cells\'celltype.loc[celltype[\'ClusterID\'].isin([\'17\']), \'celltype\'] = \'VSMCs\'# 接下来,将celltype信息映射回adata.obs# 先确保adata.obs里的cluster列也是字符串形式adata.obs[\'leiden_1.0_str\'] = adata.obs[\'leiden_1.0\'].astype(str)# 建立映射字典:cluster_id -> celltypemapping = dict(zip(celltype[\'ClusterID\'], celltype[\'celltype\']))# 用映射字典给每个cell添加celltype信息adata.obs[\'celltype\'] = adata.obs[\'leiden_1.0_str\'].map(mapping)# 查看一下分布print(adata.obs[\'celltype\'].value_counts())#celltype#T/NK cells 8917#B cells 3685#Mono/Macro cells 2502#Plasma 1774#Malignant cells 809#Fibroblast 698#Mast cells 477#VSMCs 290#Eendritic cells 258#Name: count, dtype: int64width_cm = 10height_cm = 10plt.figure(figsize=(width_cm / 2.54, height_cm / 2.54))fig = sc.pl.umap( adata, color=[\"celltype\"], legend_loc=\"on data\", frameon=True, size=8, # 调整点大小,可根据需要改 legend_fontsize=12, # 调整图例字体大小 show=False)# 遍历所有文本对象,调节字体大小for txt in fig.axes.texts: txt.set_fontsize(8) # 你想要的字体大小plt.savefig(\"celltype.pdf\",bbox_inches=\'tight\') plt.show()
简单可视化
width_cm = 30height_cm = 10plt.figure(figsize=(width_cm / 2.54, height_cm / 2.54))sc.pl.dotplot(adata, marker_genes, groupby=\"celltype\", standard_scale=\"var\", show =False )plt.savefig(\"celltype_marker.pdf\",bbox_inches=\'tight\')plt.show()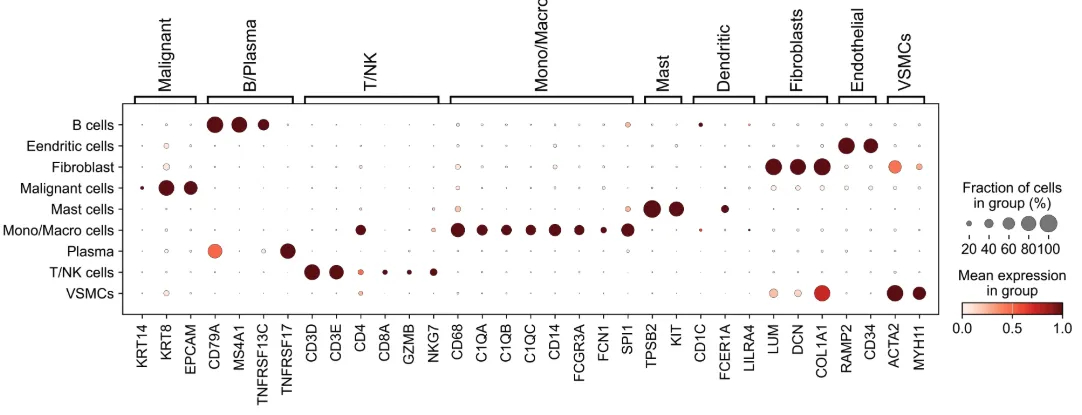
width_cm = 30height_cm = 10plt.figure(figsize=(width_cm / 2.54, height_cm / 2.54))sc.pl.stacked_violin(adata, marker_genes, groupby=\"celltype\")plt.savefig(\"celltype_marker2.pdf\",bbox_inches=\'tight\',dpi=300)plt.show()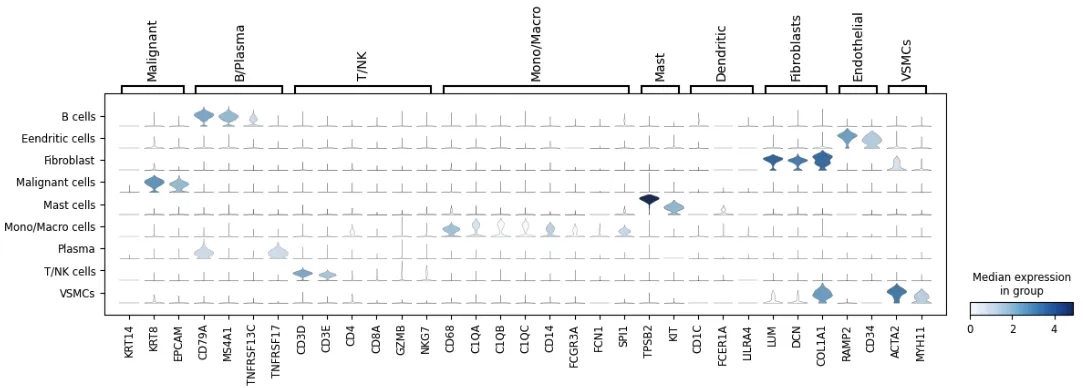
# 对celltype做一下差异分析sc.tl.rank_genes_groups(adata, groupby=\"celltype\", method=\"wilcoxon\")width_cm = 30height_cm = 10plt.figure(figsize=(width_cm / 2.54, height_cm / 2.54))sc.pl.rank_genes_groups_dotplot( adata, groupby=\"celltype\", standard_scale=\"var\", n_genes=5, show =False)plt.savefig(\"celltype_DEGs.pdf\",bbox_inches=\'tight\',dpi=300)plt.show()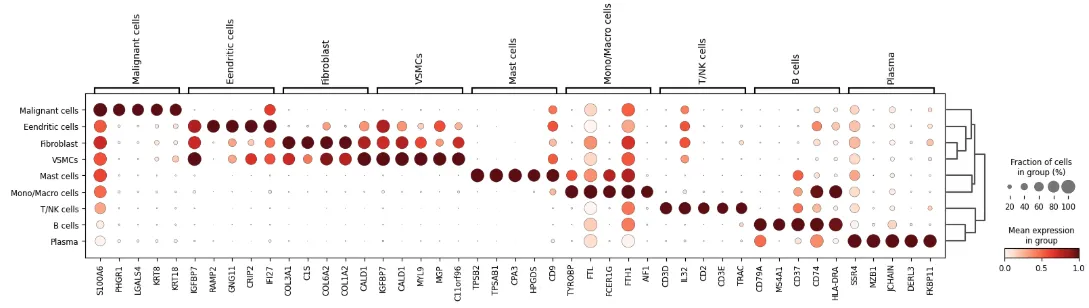
# UMAP基因图width_cm = 30height_cm = 10plt.figure(figsize=(width_cm / 2.54, height_cm / 2.54))sc.pl.umap(adata, color=[\"CST3\", \"NKG7\", \"CD3E\"], show =False)plt.savefig(\"genes.pdf\",bbox_inches=\'tight\',dpi=300)plt.show()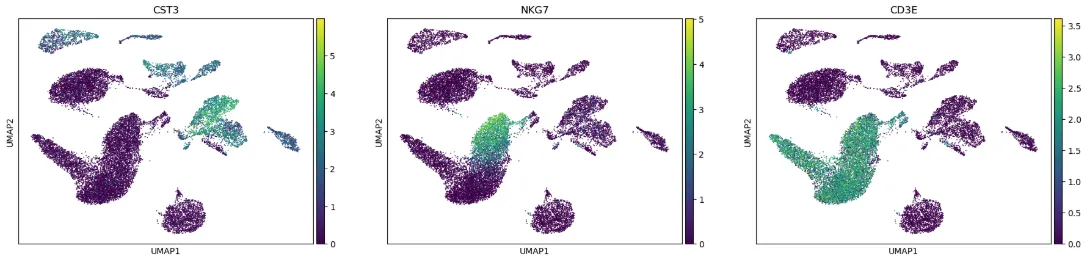
# 基因-小提琴图width_cm = 60height_cm = 10plt.figure(figsize=(width_cm / 2.54, height_cm / 2.54))sc.pl.violin(adata, [\"CST3\", \"NKG7\", \"CD3E\"], groupby=\"celltype\", show = False)fig = plt.gcf() # 获取当前figure# 遍历所有文本对象,调节字体大小for ax in fig.axes: for label in ax.get_xticklabels(): label.set_rotation(45) label.set_fontsize(12)plt.savefig(\"ViolinPlot.pdf\",bbox_inches=\'tight\',dpi=300)plt.show()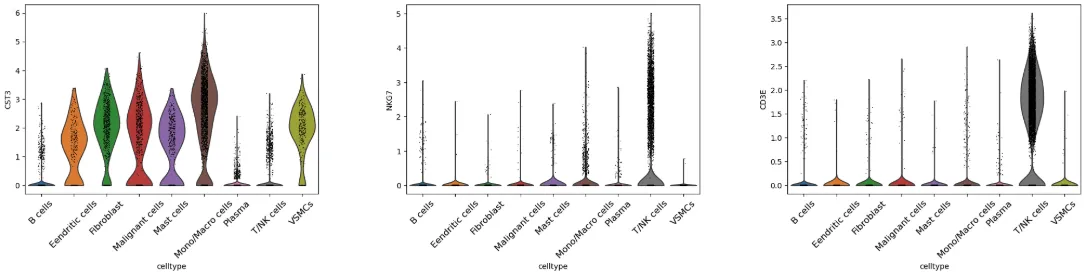
保存数据
results_file = \"./sce.all.h5ad\"adata.write(results_file, compression=\"gzip\")以上是Scanpy的完整分析流程。接下来有一个问题:在Scanpy中,如何实现类似Seurat中FindSubCluster函数的功能?
参考资料
-
scanpy:https://scanpy.readthedocs.io/en/stable/
-
scanpy中多种数据整合方式:https://nbisweden.github.io/workshop-scRNAseq/labs/scanpy/scanpy_03_integration.html
致谢:感谢曾老师以及生信技能树团队全体成员。更多精彩内容可关注公众号:生信技能树,单细胞天地,生信菜鸟团等公众号。
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多相关内容可关注公众号:生信方舟。