通过小样本学习利用少量湿实验数据提升蛋白质语言模型效率

摘要
精确建模蛋白质适应度景观 (protein fitness landscapes)对蛋白质工程至关重要。预训练蛋白质语言模型在无需湿实验数据的情况下已实现顶尖水平的蛋白质适应度预测,但其准确性和可解释性仍有限制。而传统监督深度学习模型需大量标记训练样本实现性能提升,存在实际应用壁垒。本研究提出FSFP训练策略,能在极端数据稀缺条件下高效优化蛋白质语言模型的适应度预测。该策略融合元迁移学习、排序学习与参数高效微调技术,仅需目标蛋白的数十个标记单点突变体即可显著提升多种蛋白质语言模型性能。基于87个深度突变扫描数据集的算法基准测试表明,FSFP在无监督与监督基线模型中均具显著优势。此外,我们通过湿实验成功应用FSFP改造Phi29 DNA聚合酶,阳性率提升达25%。这些结果印证了该方法在基于AI的蛋白质工程领域的应用潜力。
背景介绍
蛋白质在生命活动中不可或缺。因其具备绿色、高效、经济的生物催化剂特性,科研与工业生产中对蛋白质应用的需求持续增长。然而,多数直接从生物物种获取的野生型蛋白无法直接应用于工业环境,其部分理化性质(如稳定性、活性和底物特异性)尚不理想。蛋白质工程致力于挖掘具有特定应用价值的蛋白质。传统蛋白质工程通过定向进化和理性设计等方法力求提高这些性质。尽管定向进化效果显著,但因高通量检测存在搭建复杂度高、成本高的限制,其在筛选海量突变体文库时面临挑战。理性设计虽降低实验需求,却常因缺乏详尽结构知识与机理认知而受限。近年来,深度学习在揭示蛋白质序列与其功能(即适应性)的隐式关联方面展现出巨大潜力,为高效探索广阔设计空间提供助力。
通常,深度学习方法可分为监督与非监督模型,主要区别在于训练数据是否需要人工收集的标签。预训练蛋白质语言模型(PLMs)是当前最热门的非监督适应度预测方法。诸如ESM-2、ProGen、SaProt和ProtT5等模型,经广阔蛋白质宇宙训练后,可独立于实验数据估算各种蛋白序列的概率分布。该能力虽能预测突变效应,但准确性有限。由于这些模型本质上表征天然蛋白质序列的统计特征,其针对突变适应度的零样本似然评分实则是衡量突变蛋白序列与天然蛋白质或特定蛋白家族的相似度。尽管该指标可预测某些天然蛋白质特性(如溶解性与稳定性),却无法预测非天然催化特性,例如非天然底物的催化或非天然产物的生成。
相比之下,监督深度学习模型近期在预测蛋白质适应度方面展现出高精度特征。凭借其提取蛋白质局部与全局特征的强大能力,这些模型可通过训练充足标记数据构建更精确的序列-适应度关联。然而,它们严重依赖源自昂贵的高通量诱变实验的海量数据,对多数蛋白质构成重大挑战。近期Hsu等人开发了高效岭回归模型,融合氨基酸独热编码特征及经无监督模型计算所得的概率密度特征。在有限标记数据训练时,该模型较复杂昂贵方法表现出性能提升。但独热编码特征信息量不足,难以表征不同残基间关系;且作为线性模型,岭回归可能难以学习影响蛋白质适应度的复杂模式。因此,开发新策略以利用稀缺湿实验数据在蛋白质工程中高效微调蛋白质语言模型具有重要价值,可实现无监督与监督方法优势的融合。
本工作融合元迁移学习(MTL)、排序学习(LTR)和参数高效微调的协同方法,开发出训练PLMs的通用策略FSFP(蛋白质适应度预测的小样本学习,Few-Shot Learning for Protein Fitness Prediction)。该策略以依赖目标蛋白的极简标记数据集为特点——仅需数十个随机单点突变体。通过FSFP,此类精简数据可显著提升训练模型预测突变效应的准确性。为验证方法,我们使用ESM-1v、ESM-2和SaProt等代表性PLMs进行算法基准测试。FSFP虽理论兼容所有蛋白语言模型(PLMs),但模型选择受计算效率与资源限制等实际因素主导。在包含87个深度突变扫描(DMS)数据集ProteinGym的基准测试中,本方法展现出卓越性能,且适配不同PLMs与蛋白时具有鲁棒性,其表现优于基于数十条数据训练的无监督与监督模型。特别指出,使用目标蛋白20个标记单点突变体时,FSFP将PLMs的斯皮尔曼相关系数平均提升高达0.1。此外,湿实验应用FSFP改造Phi29 DNA聚合酶,使基于ESM-1v的前20位预测平均熔解温度(Tm)与阳性率同步提升。这些结果印证其数据利用效率,彰显其助力AI引导蛋白质工程的潜力。
结果
通过FSFP在有限训练数据下迁移PLMs
FSFP利用元学习优化标签稀缺场景的PLMs训练。元学习旨在通过积累多任务学习经验,训练出仅需少量样本和迭代即可快速适应新任务的模型。为构建元学习所需训练任务,我们检索现有标记突变体数据集(可辅助预测目标蛋白变异效应),同时通过多序列比对(MSA)为候选突变体生成伪标签(图1a及“方法”部分)。此阶段中,目标蛋白与数据库中的野生型序列或结构首先由PLMs编码为嵌入向量。选用ProteinGym作为检索数据库,因其是当前最大的DMS数据集公共资源库。随后,选取向量空间最接近目标蛋白的前两种蛋白关联数据集构建前两个任务——相似蛋白的适应度景观可能具共性。现有文献表明利用MSA预测遗传变异效应具有潜力,故采用基于比对的方法GEMME整合目标蛋白MSA信息,对目标候选突变体评分以构建第三任务数据集。各任务标记数据随机划分为训练集与测试集。由此期望元训练模型能融合进化信息与相似适应度景观来利用目标训练数据。
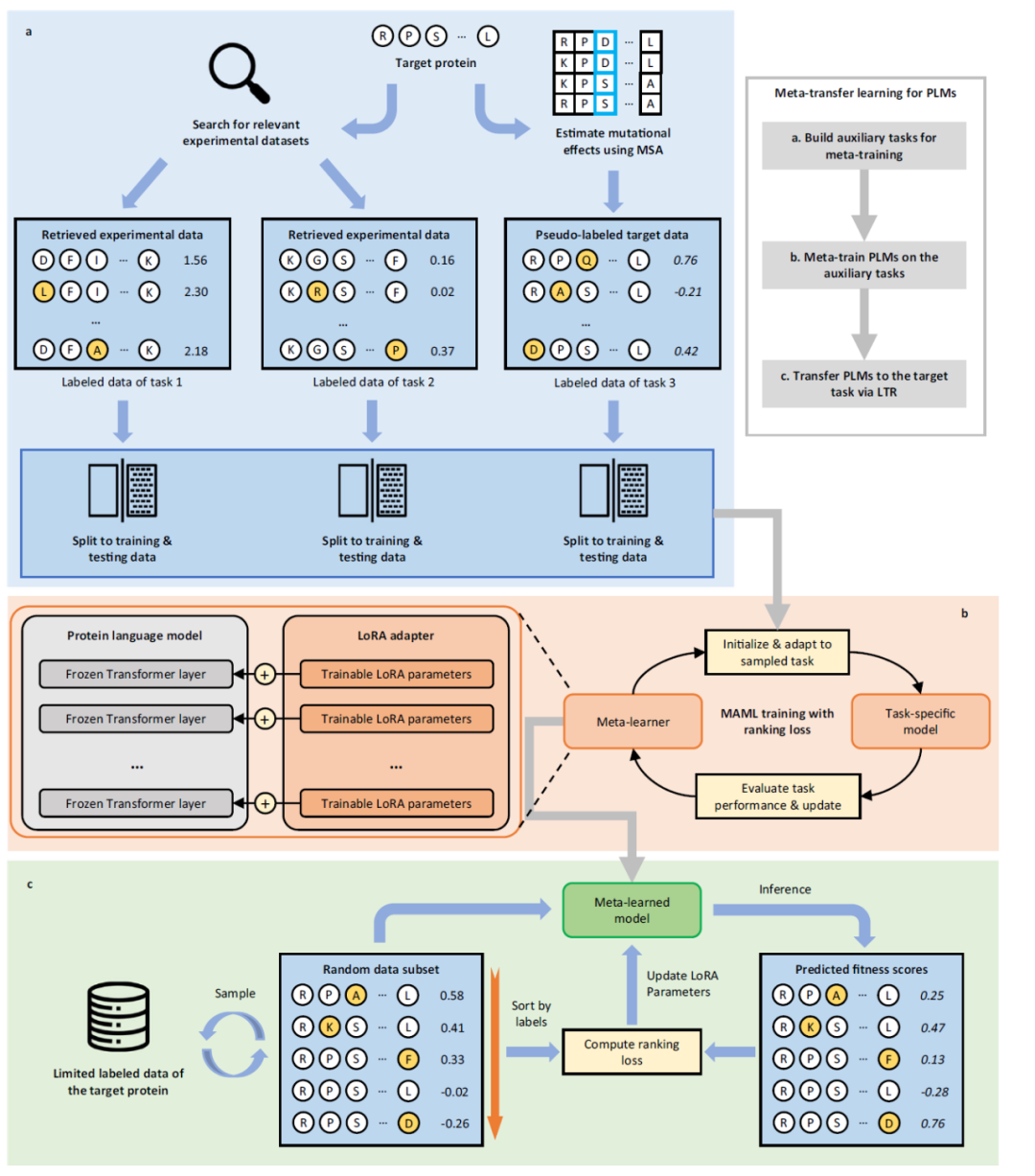
图1 | FSFP流程总览。FSFP包含三阶段:构建元学习辅助任务、在辅助任务上元训练PLMs、迁移PLMs至目标任务。a 基于目标蛋白野生型序列或结构,检索两种相似蛋白的标记突变体数据集构建前两个任务;同时采用基于MSA的方法预估候选突变体变异效应作为第三任务伪标签。b 应用MAML算法在构建任务上元训练PLM,最终将其优化为基于目标任务微调后的良好参数初始化的元学习器(右)。为防PLMs在小训练数据上过拟合,采用LoRA将模型更新约束至有限参数(左)。c 将元训练模型迁移至目标小样本学习任务。FSFP将适应度预测视为排序问题,在迁移学习和元训练中均应用LTR技术,通过计算预测值与真实排序间的列表排序损失训练PLMs排序适应度。
我们采用基于梯度的流行元学习方法——模型无关元学习 (MAML, model-agnostic meta-learning ),在构建的任务上对PLMs进行元训练(图1b及“方法”部分)。本质上,MAML通过寻找最优初始模型参数,使参数的微小调整即可显著提升目标任务性能。元训练过程每次迭代含两层优化,最终将PLM转化为用于初始化的元学习器(图1b右)。内层优化中,当前元学习器初始化临时基础学习器,再通过采样任务的训练数据将其更新为任务专用模型;外层优化中,该任务专用模型在对应任务上的测试损失被用于优化元学习器。
PLMs通常采用高参数量Transformer模型作为基础架构,并基于大规模未标记蛋白序列或结构进行预训练。但在极少标记训练数据上微调时,易遭遇灾难性过拟合。为此,FSFP应用低秩自适应(LoRA)技术,向PLMs注入可训练秩分解矩阵并冻结原始预训练参数,所有模型更新均约束于这些少量可调参数(图1b左及“方法”部分)。
元训练后,即可获得LoRA参数的好的初始化结果,最终将元训练PLMs迁移至目标小样本学习任务——即利用有限标记数据预测目标蛋白的突变效应。传统监督蛋白质适应度预测方法将此视为回归问题,而FSFP将其作为排序问题处理并采用LTR技术(图1c及“方法”部分)。蛋白质工程与定向进化中,关键指标在于突变是否增强现有蛋白的功能适应性。因此相较于关注突变的精确得分值,其相对效能或排序位次更具意义。具体而言,FSFP通过计算ListMLE损失学习适应度排序,该损失由”正确排序的置换似然度”定义。每次迭代中训练模型修正其对抽样数据子集的预测,以逼近真实排序置换。此训练方案同步应用于迁移学习阶段(使用目标训练数据)和元训练阶段的内层优化过程(使用辅助任务训练数据)。
基准测试
我们在ProteinGym替换基准上评估模型性能,该基准包含87项DMS检测中约150万错义变异。其中11个数据集含多位点突变体。ProteinGym原用于评估PLMs零样本性能,现将其转为小样本学习基准:针对每个数据集,先随机选择20个单点突变体作初始训练集,再采样另20个单点突变体将训练集扩增至40个。据此分别构建60、80和100条数据的训练集。每个训练集对应使用剩余全部数据(或部分指定数据)作为测试集,并通过训练数据交叉验证确定训练超参数(”方法”部分)。数据划分过程采用不同随机种子重复五次,取特定训练规模下各划分的平均性能。大多数实验中,预测性能采用斯皮尔曼秩相关性和归一化折损累积增益(NDCG, normalized discounted cumulative gain)两项指标,以适应度标签为基准进行衡量。
NDCG具体见:https://zhuanlan.zhihu.com/p/371432647
在搜索和推荐任务中,系统常返回一个item列表。如何衡量这个返回的列表是否优秀呢?
Gain: 表示一个列表中所有item的相关性得分
Cumulative Gain: 对列表中每个item的Gain进行累加
Discounted Cumulative Gain: 对列表中每个item的Gain除以其排序位置系数后进行累加,使得排序靠前的权重大,排序靠后的权重小
Normalized Discounted Cumulative Gain: 根据理想排序的DCG值进行归一化后的结果
小样本蛋白质适应度预测的主要基线为Hsu等人提出的岭回归方法。该方法融合位点特异性残基的独热编码特征与现有进化概率密度模型预测的适应度得分作为输入特征训练岭回归模型。该模型虽结构简单,但在资源匮乏场景下比其他监督学习方法更有效。将FSFP应用于基础模型评估时,我们将其与经岭回归增强的该模型版本对比,并使用该基线的官方实现以确保性能可靠性。
FSFP每个步骤对小样本学习均具正向贡献
为全面评估FSFP各步骤的影响,我们以ESM-2为基础模型进行消融研究。选定650 M参数规模(其他规模亦经评估,FSFP在更大模型上持续表现更优,见补充图1a)。具体对比策略如下:
(1) LTR + LoRA + MTL (无MSA):FSFP变体,不依赖MSA构建辅助任务,改用数据库检索的标记数据集替代第三任务
(2) LTR + LoRA:FSFP变体,通过LTR将LoRA适配模型迁移至目标任务,未在辅助任务上元训练
(3) MSE:按Rives等方法微调整个PLM,以突变体与野生型的对数似然差为预测指标
(4) MSE + LoRA:用LoRA增强的MSE方法
(5) 岭回归:Hsu等人提出的方法
(6) 零样本推断:按Meier等人方法使用PLM进行零样本预测
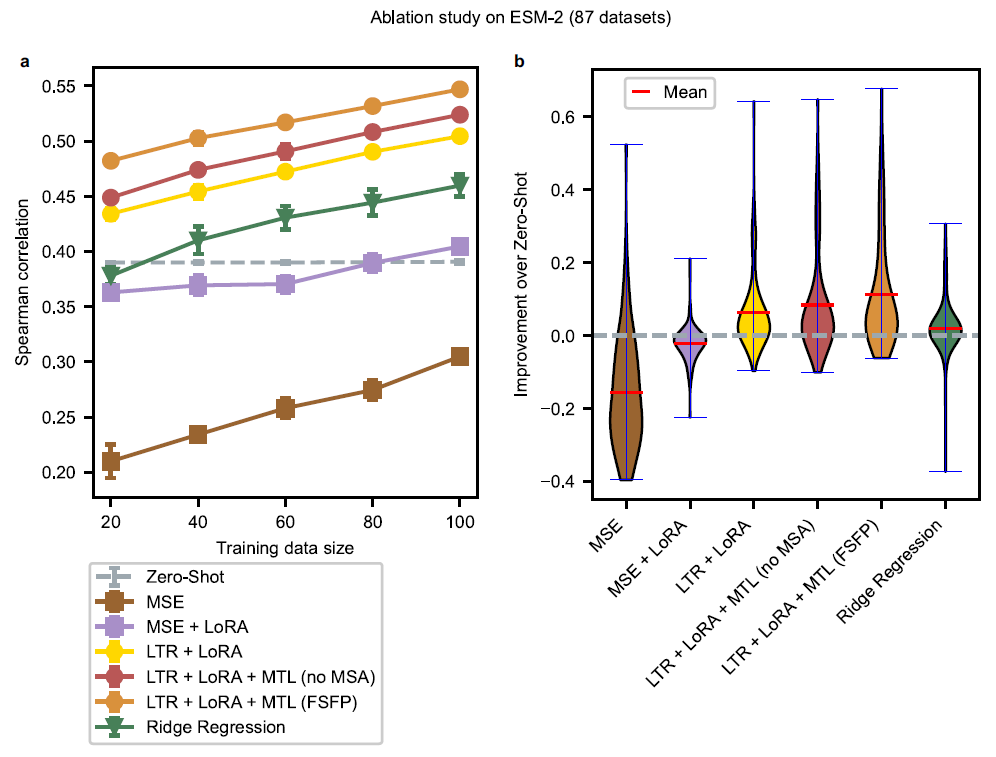
图2 | ESM-2消融研究。a 各策略在ProteinGym所有数据集上随训练数据量变化的平均性能(斯皮尔曼相关性评估)。每个数据集随机选取少量(20、40、60、80、100)单点突变体作为训练集,其余为测试集。图中每个点为五次随机数据划分的平均测试性能,误差棒指示不同划分导致的标准差。采用双尾曼-惠特尼U检验 (Mann–Whitney U tests)比较FSFP与其它策略性能,所有训练规模中最大P值为0.0079。NDCG、皮尔逊相关性与MAE的等效结果见补充图1b-d。b 训练规模40时,ProteinGym所有数据集上斯皮尔曼相关性较零样本预测的性能提升分布。各数据集性能增益取五次随机划分的平均值。源数据详见源数据文件。
相较于零样本推断,MSE方法导致平均预测性能显著下降(图2a及补充图1b,c)。这归因于小训练数据集上直接微调整个模型引发的快速过拟合(补充图2b,c)。引入LoRA通过高效参数化实现最小化调整,整体显著缓解此类负面效应,但其性能直至训练集达80条数据仍未能匹配原始无监督模型。作为主要基线,岭回归在多数训练规模下性能超越零样本推断,与Hsu等人的对比结果一致。LTR + LoRA在所有训练规模上均优于上述方法(图2a及补充图1b,c)。同时,在大多数数据集上,LTR较回归方法大幅提升模型性能(图2b)。如前所述,定向进化中不同突变体的排序位次常比绝对分值更重要。此外,小样本学习场景下,因训练与测试数据的标签值范围通常存在显著差异,精确预测具体标签值极具挑战。实际上,当引入平均绝对误差(MAE)评估时,基于回归的方法表现同样不佳(补充图1d),致使其绝对值输出缺乏应用价值。由于排序位次更具实际意义,适应度预测优先采用排序相关指标,故LTR技术更为适用。
通过MTL获取小样本训练的初始模型参数可进一步提升性能(图2a及补充图1b,c)。LTR + LoRA + MTL(无MSA)相较LTR + LoRA的改进表明,其他蛋白适应度景观的相似特性具有参考价值。FSFP在元训练中额外学习MSA知识,所有训练数据规模下均获最优评分(图2a及补充图1b,c),这提示除目标蛋白训练数据外,MSA提供的进化信息也可有效指导模型估计突变效应。如训练曲线所示(补充图2),无论是否含MSA,MTL较其他方法能以极少训练迭代显著提升模型性能。此外,经元训练的初始模型在未接触目标训练数据(即零训练迭代)时,某些情况下已然显著超越零样本推断。这些证实应用FSFP可使模型成功从辅助任务中学习有效信息,从而良好迁移至目标小样本学习任务。
FSFP作为PLMs通用小样本学习方法
FSFP适用于任何基于梯度下降优化的深度学习蛋白质适应度预测器,本研究聚焦PLMs。为验证普适性,选取三种代表性PLMs——ESM-1v、ESM-2和SaProt——作为训练基础模型(“方法”部分)。均采用650M版本评估,其中可训练LoRA参数占模型总量的1.84%。在全部87个基准数据集上,将FSFP与零样本预测及岭回归方法对比。因FSFP利用GEMME生成元训练伪标签,另增GEMME和经岭回归增强的GEMME作为额外基线。单点与多位点突变体的测试性能分别报告(图3及补充图3)。需说明,Hsu等人报告的最佳方法是经岭回归增强的DeepSequence,但其在ProteinGym整体性能弱于经岭回归增强的GEMME,故图中未纳入此法。
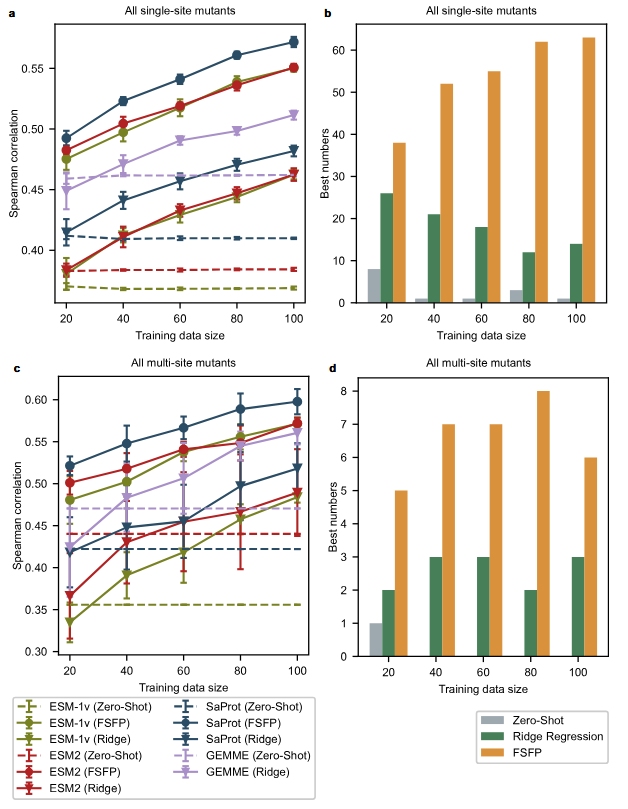
图3 | 单点与多位点突变预测性能综合比较。a 全部87个数据集单点突变体测试平均性能(斯皮尔曼相关性评估)。误差棒表示五次随机划分导致的标准差。SaProt(FSFP)显著优于所有基线,所有训练规模中最大P值为0.0079(双尾曼-惠特尼U检验)。NDCG等效结果见补充图3a。b 各数据集单点突变体最佳斯皮尔曼相关性由PLM实现的频次统计,颜色代表应用于最优PLMs的不同策略。c 11个数据集多位点突变体测试平均性能(斯皮尔曼相关性评估)。误差棒表示五次随机划分导致的标准差。SaProt(FSFP)显著优于所有基线,所有训练规模中最大P值为0.016(双尾曼-惠特尼U检验)。NDCG等效结果见补充图3b。d 类同(b)但统计多位点突变体最佳性能。源数据详见源数据文件。
就平均性能而言,经FSFP训练的PLMs在所有训练数据规模下持续优于其他基线(图3a,c)。其中SaProt(FSFP)表现最佳,ESM-1v(FSFP)与ESM-2(FSFP)性能相当。此外,ProteinGym多数数据集上的最佳斯皮尔曼相关性均由FSFP训练后的PLMs实现(图3b,d)。相较零样本预测,FSFP仅用20个训练样本即令PLMs在单点突变体上的斯皮尔曼相关性提升近0.1,而该差距在多位点突变体预测中进一步扩大。随着训练集扩展,性能改进持续增强,与先前消融研究结果一致,证实FSFP对不同基础模型的普适性与有效性。相比之下,岭回归方法在20个训练样本时未能显著超越对应零样本预测(图3a,c)。对于多位点突变体,训练规模为20时该方法对GEMME、ESM-1v和ESM-2产生严重负面效应。尽管其性能随训练规模增大而提升,但始终落后于FSFP训练模型。除训练策略差异外,这很可能源于岭回归方法中简单独热特征的表征局限及模型容量不足。需特别指出,预测多位点突变体时FSFP的标准误差远小于岭回归,表明前者在不同小样本训练数据下更具稳定性与可靠性。
值得注意的是,GEMME的平均零样本性能优于所选PLMs。这并不意外——尽管PLMs对蛋白质序列或结构统计特征建模能力强大,但其预测变异效应的表现未必始终优于基于MSA的方法。因此在如此小样本场景下,参考前沿MSA方法的指导具有合理性。如图3a所示,采用FSFP的模型在所有训练规模上均较GEMME及其岭回归增强版实现显著提升。这表明FSFP不仅将GEMME的MSA知识注入PLMs,更通过MTL成功将其与目标训练数据的监督信息融合。这再次验证了FSFP作为小样本学习策略的优越性,尤其在训练数据集极其有限时。
FSFP具备强泛化能力与外推性
蛋白质工程中,未见于标记数据的突变位点的效应始终受关注。因此,适应度预测器的位点外推能力对从海量序列空间发掘优良突变体至关重要。我们通过评估测试数据特定子集检验不同方法的外推能力:从原始测试集先筛选突变位点区别于训练样本的全部单点突变体,构成更高难度单点测试集;再选择单个突变与训练数据无重叠的多位点突变体,形成另一挑战性测试集。未强制测试多位点突变体的所有突变位点均非训练位点,因为这将导致部分数据集测试样本不足。此设定下可见基础模型的零样本性能随训练集规模明显波动(图4),归因于测试集剧变——训练集扩增使符合上述条件的可用测试样本在某些数据集中急剧减少,影响零样本预测评估结果。
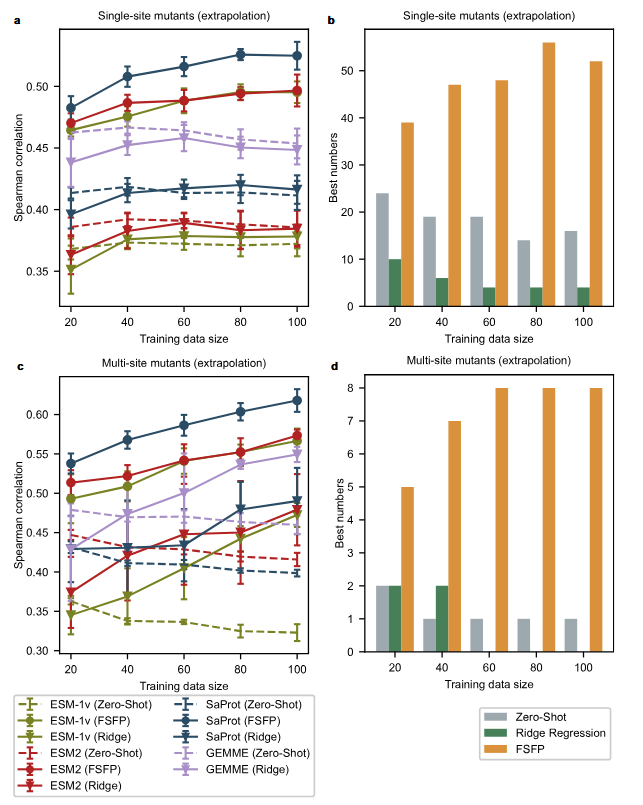
图4 | 单点与多位点突变体的外推性能。a 外推至突变位点未出现于训练集的单点突变体(斯皮尔曼相关性评估)。误差棒以平均性能为中心,表示五次随机划分导致的标准差。SaProt(FSFP)显著优于所有基线,所有训练规模中最大P值为0.016(双尾曼-惠特尼U检验)。NDCG等效结果见补充图4a。b 各数据集单点突变体最佳外推斯皮尔曼相关性由PLM实现的频次统计,颜色代表应用于最优PLMs的不同策略。c 外推至单个突变与训练数据无重叠的多位点突变体(斯皮尔曼相关性评估)。误差棒以平均性能为中心,表示五次随机划分导致的标准差。SaProt(FSFP)显著优于所有基线,所有训练规模中最大P值为0.0079(双尾曼-惠特尼U检验)。NDCG等效结果见补充图4b。d 类同(b)但统计多位点突变体最佳外推性能。源数据详见源数据文件。
外推至不同位点的单点突变体时,即使使用100个训练样本,经岭回归增强的模型仍未见明显优于基础模型(图4a)。对于多位点突变体,当训练规模<60时,岭回归方法未能有效提升GEMME和ESM-2性能,且如前述表现出比FSFP更大的标准差(图4c)。与之形成鲜明对比,经FSFP训练的PLMs在不同训练规模下持续以更高斯皮尔曼相关性评分超越所有基线。相较其零样本性能,经FSFP训练后获得显著提升(ESM-1v尤为突出),且多数数据集的最佳预测器均为FSFP训练模型(图4b,d)。该结果与图3量化结论一致:PLMs凭借预训练赋予的高容量与内嵌知识具备强泛化性,可通过恰当训练范式提升困难下游任务表现。然而岭回归方法的输入特征信息量不足,致使其外推能力受限。
为深入验证FSFP的适用性与泛化能力,我们在四种蛋白上展示不同方法的结果比较:HIV包膜蛋白Env、人α-突触核蛋白、G蛋白(GB1)及人TAR DNA结合蛋白43(TDP-43)。这些案例中部分无监督模型表现欠佳且实践可靠性不足,凸显利用有限标记数据有效训练的必要性(补充图5-6)。值得注意的是,TDP-43实验中所有模型零样本结果与实际适应度标签无相关甚至负相关(补充图5d);ESM-2在HIV Env(补充图5a)和α-突触核蛋白(补充图5b)上准确性低下。可见除GB1外(补充图5c),大多数岭回归增强模型即使使用更大训练集也较零样本性能未显显著改进。相反,FSFP赋能PLMs通过小数据集训练获得显著提升,在定向进化中更具实用性。此外可观察到,FSFP生成伪标签所用的GEMME在后三例中性能并未领先其他基础模型,但这不妨碍FSFP训练模型成为最优预测器,表明FSFP从辅助任务中学习泛化能力而非简单过拟合。
FSFP在Phi29 DNA聚合酶工程化中的应用
我们通过湿实验验证FSFP的实际效能:改造Phi29 DNA聚合酶(”方法”部分)。该聚合酶在生物技术应用中有关键作用,已被充分验证为高效等温DNA扩增酶。提升Phi29热稳定性是当前研究热点。本研究通过先获取足量阳性单点突变体增强其热稳定性,继而衍生潜在更优多位点突变体。基于有限湿实验数据应用FSFP训练ESM-1v,进而筛选新型单点突变体进行实验验证(图5a及”方法”部分)。
最初在无湿实验数据时,基于ESM-1v零样本预测筛选Phi29前20位单点突变体进行首轮湿实验。构建并纯化这些突变体后测定其热稳定性,所测Tm值与野生型基准值对比。随后以全部20个突变体的Tm值作为标签,通过FSFP训练ESM-1v。经优化的模型进一步预测新一轮前20位单点突变体用于深化湿实验。
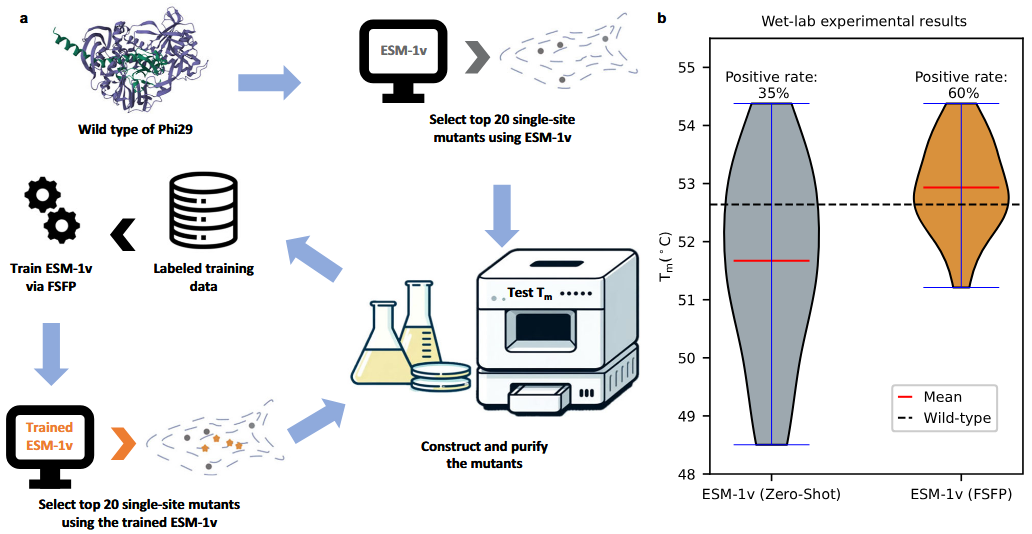
图5 应用FSFP改造Phi29。a FSFP改造Phi29 DNA聚合酶的工作流程。b ESM-1v经FSFP训练前后预测所得前20位单点突变体的湿实验Tm值。源数据详见源数据文件。
比较ESM-1v经FSFP训练前后的前20位预测结果,可见平均Tm值提升超1°C,阳性率提高25%(图5b及补充表1)。具体而言,ESM-1v(FSFP)发现的Tm值最优突变体同样被ESM-1v(零样本)推荐;但在其预测的阳性突变体中,九个未出现于训练数据,表明FSFP可使PLMs识别出更多优于野生型的蛋白质变体。这些结果证实FSFP在加速蛋白质工程设计-测试迭代循环中的潜力,有助于开发具增强功能特性的蛋白质。
讨论
本研究提出FSFP范式,能利用少量(数十个)标记突变体有效训练蛋白质语言模型预测适应度。该范式融合了LTR、LoRA与MTL技术:LTR满足定向进化对蛋白质适应度排序的内在需求,LoRA极大降低小训练集导致的过拟合风险,MTL提供更优初始化参数实现快速蛋白质适配。案例研究应用于三种代表性模型(ESM-1v/ESM-2/SaProt),但该方法理论上兼容所有蛋白质语言模型。通过87个深度突变扫描数据集的综合计算实验,验证FSFP在小样本蛋白质适应度预测中的有效性与稳健性:(1)仅用20训练样本即可提升模型斯皮尔曼相关性达0.1;(2)不同模型性能持续显著提升;(3)能精准预测训练数据未覆盖的突变位点;(4)零样本性能较弱时仍保持高效数据利用。湿实验应用FSFP改造Phi29 DNA聚合酶证实:ESM-1v预测的前20位突变体平均熔解温度与阳性率均显著提升。
符合常理的,当元训练使用的蛋白质包含更多突变体且与目标蛋白质相似度更高时,迁移学习性能显著提升(附图7a)。相较于无MTL的微调方法(LTR+LoRA),在辅助任务数据集规模≥500时,即使检索到的蛋白质相似度较低,元学习仍能发挥积极作用。由于第三项辅助任务完全基于目标蛋白质的多序列比对(MSA)构建,可有效缓解低相似度蛋白质的负面影响。值得注意的是,在最差情况下(附图7a最左侧柱图),FSFP性能仍与LTR+LoRA相当,且大幅超越零样本预测能力。其根本原因在于:1)利用目标训练数据实施早停策略(见”方法”章节),避免模型在低质量辅助任务上过拟合;2)辅助任务所含目标蛋白质信息越丰富,元学习效果越显著。因此,基于领域知识(如采用目标蛋白不同类型实验数据)收集高关联性辅助数据集至关重要。
FSFP通过计算蛋白质语言模型生成的嵌入向量间余弦相似度搜索相关蛋白,也可采用MMseqs2、Foldseek等其他方法(附图7b-d)。总体而言,这些搜索方法的性能差异不大(附图7c-d),均能可靠识别关联训练数据集。不同模型在零样本预测中表现各异:如ESM-2在预测酶活性时最优,而SaProt在预测表达水平时领先(附图7c)。经FSFP训练后这种跨数据集性能趋势仍然存在,表明该方法在提升模型精度时可能保留其固有优势或数据偏好。由于不同模型学习各类蛋白质适应度景观时存在差异(实验中各数据集最优模型均不同),选择待训练的基础模型需谨慎考量。
基于显著的小样本性能优势,FSFP能实现更高效的定向进化,尤其当高通量筛选难以实施时。定向进化的初始数据可源于理性设计、随机突变或PLM的零样本预测。无论初始数据集中阳性突变体比例如何,均可据此筛选最适用的PLM。在后续迭代循环中,可应用FSFP训练所选模型,进而利用其外推能力推荐新突变体。
方法
蛋白质适应度排序问题的高效解决方案


在每轮训练迭代中,我们从训练数据中有放回地随机抽取m个大小为n的子集(n小于训练数据规模,m可视作批大小),并对每个子集计算ListMLE损失。通过梯度下降法,使用这m个子集的平均损失更新PLM中的可训练参数。具体参数m和n的取值取决于实际训练数据规模及其在训练集上的验证性能。
元学习辅助任务
元学习旨在通过积累多任务学习经验,训练出能仅用少量数据和训练轮次快速适应新任务的模型。在将蛋白质语言模型迁移至目标小样本学习任务前,我们执行元学习以获得更优的LoRA参数初始化,从而加速目标训练数据学习并进一步降低过拟合风险。构建元学习所需训练任务时,需搜索对预测目标蛋白变异效应有益的现有标记突变数据集,以及通过多序列比对技术为候选突变体生成伪标签。
检索相似实验数据集

最终选取与目标蛋白相似度最高的两种蛋白质,其对应深度突变扫描数据集作为前两项任务的标记训练数据。
基于多序列比对的突变效应预测
研究证实多序列比对(MSA)不仅能有效预测突变效应,还可增强蛋白质语言模型的下游任务性能。本工作通过元训练将MSA知识整合至PLM:采用基于序列比对的GEMME算法生成伪标签,避免修改模型架构。该算法通过显式建模序列位点间相互依赖关系预测突变效应:利用MSA构建联合进化树,基于进化轨迹计算蛋白质序列各位置保守度,据此推演突变所需适应度。同时借助突变发生相对频率推断其效应。应用GEMME对目标蛋白候选突变序列评分,构建第三项任务数据集。
元训练蛋白质语言模型
采用前沿元学习算法MAML,使PLM能更高效利用小样本训练数据。该方法已成功应用于药物-靶点相互作用和抗原结合识别。MAML本质在于寻找最优初始参数,使其在目标损失函数驱动下微调即可显著提升任何目标任务的性能。 
基础模型
ESM1v模型:ESM-1v作为变异效应预测的Transformer语言模型,继承ESM-1b架构与Rives等的掩码语言建模方法。该模型基于Uniref90无标注蛋白序列训练,参数量达6.5亿。原始版本包含五种不同随机种子的集成模型,本实验仅采用首个检查点。
ESM-2模型:ESM-2针对ESM-1b在架构与预训练数据方面升级,采用旋转位置嵌入技术并基于UniRef50数据集训练。本实验选用其650M参数版本。
SaProt模型: Su等提出的SaProt是基于蛋白质序列与结构数据的语言模型。该模型创新引入结构感知词汇表,融合残基类型与Foldseek编码的3Di结构标记。预训练数据集包含约4000万条AlphaFold2预测结构,采用与ESM-2相同的架构,输入为结构感知标记。本研究选用基于PDB结构持续预训练的版本。针对其特殊词汇表,原评分函数(4)调整为:
早停策略
深度学习广泛采用早停法预防模型过拟合。当标记数据充足时通常基于独立验证集实施,但在低资源场景下保留验证集会加剧训练数据不足;反之若验证集过小,斯皮尔曼相关性等验证指标可能缺乏代表性。针对此矛盾,本研究提出基于蒙特卡洛交叉验证估计迁移学习训练轮次:首先创建五组训练集的随机划分(训练数据量<50时训练集与验证集比例0.5:0.5,否则0.75:0.25)。每个划分下模型在子训练集上最多训练500步,每5步记录验证集斯皮尔曼相关性。经五轮训练验证后,选取跨划分平均验证分数最高的训练步数,最终在全训练集上按此步数完成训练。

基准数据集
ProteinGym作为涵盖约150万个错义变异的综合性深度突变扫描实验平台,汇集了来自87项检测实验的数据用于比较不同突变效应预测工具。本评估采用其点突变基准数据集,针对ESM-1v模型1024氨基酸的最大输入长度限制,对长度超限的蛋白质进行截断处理并确保目标突变位点包含在截取区间内。对于SaProt模型的输入构建,通过AlphaFold2预测或直接从AlphaFoldDB数据库获取相应蛋白质结构数据。
在ProteinGym的每个数据集中,首轮随机抽取20个单位点突变体作为初始训练集。以此为基础,第二轮新增20个随机选取的单位点突变体构建扩展训练集。依此方法分阶段将训练集规模拓展至60、80及100单位点突变体进行独立实验。各训练集对应的剩余数据(或指定部分)作为测试集,全程禁止超参数选择时接触这些数据。上述数据划分流程通过不同随机种子重复五次,最终按训练规模报告平均模型性能。由于测试数据随实验变化,零样本预测性能也不恒定。
Phi29 DNA聚合酶湿实验方案
Phi29 DNA聚合酶广泛应用于滚环扩增、多重置换扩增(MDA)及非特异性全基因组扩增(WGA)等DNA扩增技术。该酶作为高效等温DNA扩增工具已被充分验证。不同扩增方法对酶学特性有差异化需求:例如在高温环境下进行的WGA与MDA反应可受益于加速的反应动力学,且提升反应温度或能减轻C/G含量对扩增偏差的影响。因此增强Phi29的热稳定性成为学术研究焦点。尽管已有大量工程化蛋白质研究,现有突变体热稳定性仍未能满足实际应用需求,迄今尚无重大突破性进展。
突变体筛选流程
初筛环节基于五模型集成的ESM-1v对Phi29单位点饱和突变体进行评分(取零样本预测均值),选取预测分最高的20个突变体实测Tm值。随后以实测Tm值为标签构建训练数据集,应用FSFP微调集成中首个ESM-1v模型优化其预测能力。经训练模型对单位点突变体的前20位预测结果,将作为下一轮湿实验的验证对象。
质粒构建
通过生工生物工程(上海)有限公司合成Phi29 DNA聚合酶及其突变体基因的密码子优化版本。将其克隆至pET28(a)质粒中(Phi29蛋白构建流程见附图8),从而构建出带有N端His标签的重组质粒pET28a-phi29-MX。
蛋白质表达
将表达质粒转化至大肠杆菌BL21(DE3)感受态细胞。取30ml种子培养液于含50 μg/ml卡那霉素的LB培养基中37℃培养,随后转移至500ml含同等浓度卡那霉素的LB摇瓶培养液。待培养液OD600值达1.0时,加入终浓度0.5mM的异丙基-β-D-硫代半乳糖苷诱导蛋白表达,并于25℃持续培养16小时。
蛋白质纯化
细胞经3345×g离心30分钟收集沉淀。沉淀重悬于裂解缓冲液(25mM Tris-HCl, 500mM NaCl, pH 7.4),使用Scientz超声破碎仪裂解。裂解液于4℃、15615×g离心30分钟,取上清液进行镍柱亲和层析,洗脱缓冲液为(25mM Tris-HCl, 500mM NaCl, 250mM咪唑, pH 7.4)。纯化蛋白使用超滤法以裂解缓冲液(25mM Tris-HCl, 500mM NaCl, pH 7.4)脱盐。含蛋白组分管于保存缓冲液(25mM Tris-HCl pH 7.4, 200mM NaCl, 20%甘油)中-20℃闪冻保存。
熔解温度测定
采用差示扫描荧光法(DSF)配合蛋白质热位移检测试剂盒(Thermo Fisher)测定Tm值。取1μL SYPRO Orange染料(SUPELCO, USA)与49μL裂解缓冲液(25mM Tris-HCl, 500mM NaCl, pH 7.4)混合制成稀释染料。将1μL稀释染料与19μL 0.1mg/mL蛋白溶液混合,使用LightCycler 480 II实时荧光定量PCR仪(Roche, USA)进行DSF检测:反应体系先平衡至25℃,再以0.05℃/秒速率升温至99℃并维持2分钟。数据处理通过Protein Thermal Shift软件完成。
数据可用性
基准测试数据集源自ProteinGym平台(https://github.com/OATML-Markslab/Tranception)。各方法在斯皮尔曼相关性与NDCG指标下的详细数据集表现分别载于补充数据1与补充数据2。本文提供相关原始数据。
代码可用性
FSFP源代码存储于https://github.com/ai4protein/FSFP.
高颜值免费 SCI 在线绘图(点击图片直达)
最全植物基因组数据库IMP (点击图片直达)

往期精品(点击图片直达文字对应教程)




























机器学习