GEO数据库分析(超详细)_geo数据分析
本文针对来自GSE205185的原发性乳腺肿瘤组织转录组数据,进行全面的生物信息学分析。首先,通过数据预处理和质量控制,确保样本的可靠性。随后,采用差异表达分析识别癌组织与正常组织中的关键基因变化,并结合功能富集分析揭示相关的生物路径。接着,利用免疫浸润分析探索肿瘤微环境中的免疫细胞组成变化,为理解乳腺癌的免疫机制提供线索。最后,通过构建协同网络,挖掘潜在的关键调控因子,为乳腺癌的诊断和治疗提供潜在的生物标志物。
一、数据获取与预处理
1.1 数据下载
本研究从NCBI GEO数据库(GSE205185)获取了原发性乳腺肿瘤组织基因表达谱数据,采用Agilent SurePrint G3 Human Gene Expression v3 8x60K Microarray芯片平台(GPL21185)。实验设计包含22组样本:17 名原发性乳腺癌 (PBC) 肿瘤组织患者和 5 名正常乳腺组织供体
setwd(choose.dir())#自行选择工作目录#加载R包####library(readxl)library(tidyverse)library(GEOquery)library(tidyverse)library(GEOquery)library(limma) library(affy)library(stringr)library(parallel)library(doParallel)library(data.table)#下载数据####GEOdata = getGEO(\'GSE205185\', destdir=\".\", AnnotGPL = T, getGPL = T)#读取平台文件####GEOGPL<- getGEO(filename =\"GPL21185.soft.gz\", AnnotGPL = T)GPLtable<- Table(GEOGPL)#提取表达量####GEOexp <- exprs(GEOdata[[1]])1.2 数据预处理
原始数据需要经过多个步骤的清洗和转换:
-
探针到基因的转换:使用平台注释文件(GPL96)将探针ID转换为基因ID
-
处理重复基因:多个探针对应同一基因时取表达量平均值
-
去除缺失值和空白基因:保证数据质量
-
处理一对多关系:去除一个探针对应多个基因(适用于WGCNA等网络分析)
probe_name<-rownames(GEOexp)#记录行名(基因ID)#转换ID####loc<-match(GPLtable[,1],probe_name)#匹配基因IDprobe_exp<-GEOexp[loc,]#提取匹配的表达量raw_geneid<-(as.matrix(GPLtable[,\"GENE_SYMBOL\"]))#提取基因名index<-which(!is.na(raw_geneid))#记录空值与非空值ID的位置geneid<-raw_geneid[index]#保留基因名以及位置信息exp_matrix<-probe_exp[index,]#提取表达量去除空值对应行##将基因名转换入表达量####geneidfactor<-factor(geneid)gene_exp_matrix<-apply(exp_matrix,2,function(x) tapply(x,geneidfactor,mean))#替换基因ID,多个探针对应一个基因取平均值,tapply 的结果会以 geneidfactor 的 level 作为行名#rownames(gene_exp_matrix)<-levels(geneidfactor)gene_exp_matrix=na.omit(gene_exp_matrix)#去除第一行空值行####1、直接删除一个探针对应多个基因(适用于WGCNA等网络分析)gene_exp_matrix <- as.matrix(gene_exp_matrix)# 保留行名中不包含 \'/\' 的行gene_exp_matrix <- gene_exp_matrix[ !grepl(\"/\", rownames(gene_exp_matrix)), ]gene_exp_matrix<-as.data.frame(gene_exp_matrix)#将矩阵转换为数据框,方便进行后续的数据处理和分析。#读取分组信息####pdata <- pData(GEOdata[[1]])group_list <- pdata$`tissue:ch1`#$在右边表示读取这一列,在左边表示创建一列group_list = factor(group_list)#将向量转换成因子pdata$group=group_list#把因子添加到pdata的最后一列#导出数据####fwrite(file = \"GEOexp.txt\", gene_exp_matrix, sep = \"\\t\", quote = F, row.names = T)fwrite(file = \"GEOpdata.txt\", pdata, sep = \"\\t\", quote = F, row.names = T)
二、数据预处理(质控)
2.1 做差异分析前还需对样本进行聚类,去除离群样本
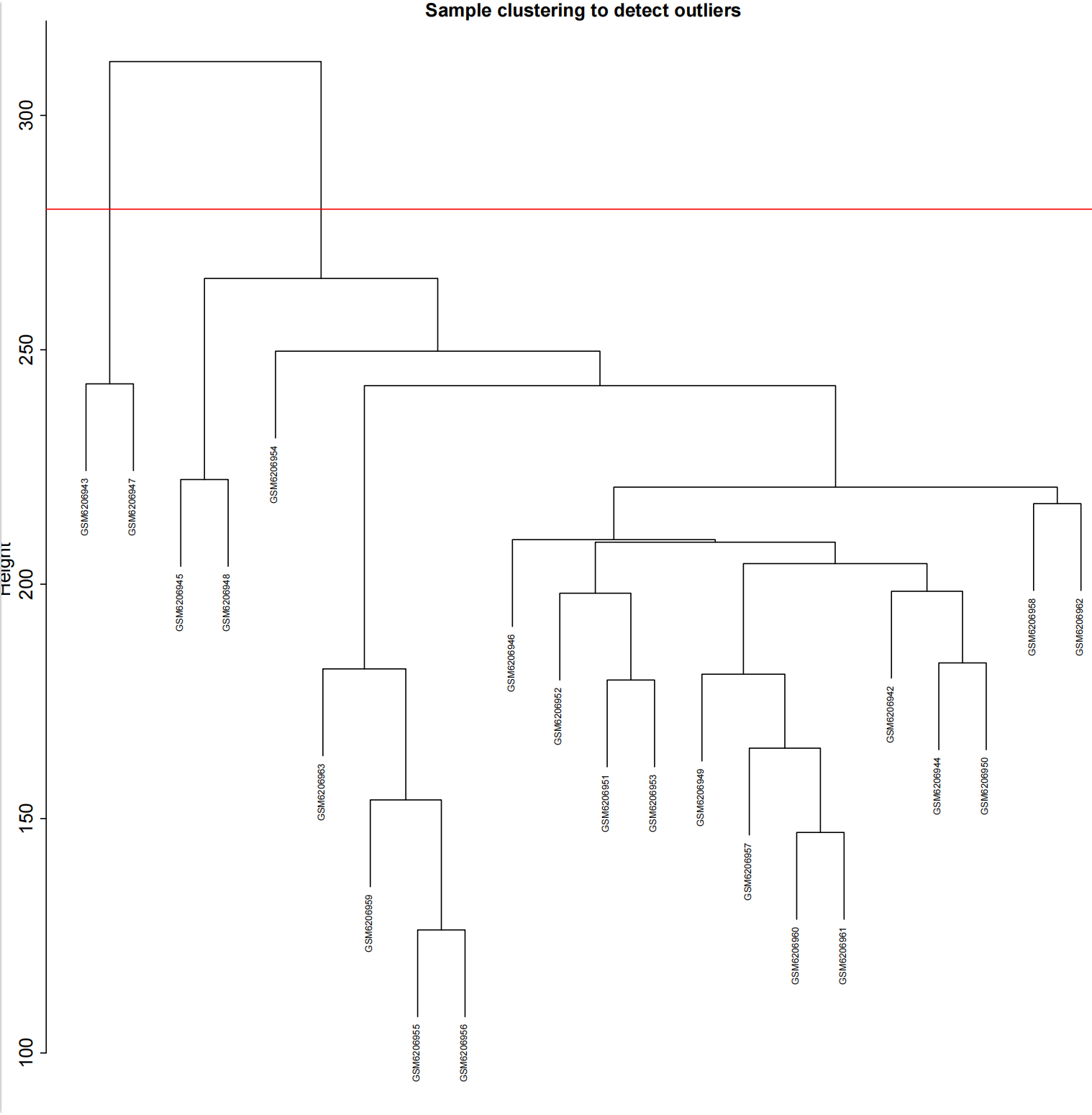
#加载R包####library(sva)library(FactoMineR)library(factoextra)library(limma)library(parallel)library(cowplot)library(WGCNA)source(\"xhhGEOQC.R\",encoding = \"utf-8\")randomColor <- function() { paste0(\"#\",paste0(sample(c(0:9, letters[1:6]), 6, replace = TRUE),collapse = \"\"))}randomColors <- replicate(10,randomColor())#读取数据####GEOdata=fread(\"GEOexp.txt\",header = T,check.names = F,data.table = F)rownames(GEOdata)=GEOdata$V1GEOdata=GEOdata[,2:ncol(GEOdata)]#读取分组####GEOgroup<-fread(\"GEOpdata.txt\",header = T,check.names = F,data.table = F)group<-data.frame(ID=GEOgroup$V1,group=GEOgroup$group)rownames(group)<-group$ID#判断GEO数据是否需要取log2xhhGEOQC(GEOdata)#进行PCA分析,查看是否有离群样本dat0=as.data.frame(t(GEOdata))dat.pca0 <- PCA(dat0, graph = FALSE)pca_plot0 <- fviz_pca_ind(dat.pca0,geom.ind = \"point\", col.ind = group$group, palette = randomColors[1:2], addEllipses = TRUE, legend.title = \"Groups\")ggsave(plot = pca_plot0,filename =\"PCA_GEO.pdf\")#进行样品聚类分群####GEOdatahclust=as.matrix(t(GEOdata))sampleTree = hclust(dist(GEOdatahclust), method = \"average\")pdf(file = \"样品聚类_GEO.pdf\", width = 20, height = 20)par(cex = 0.5)par(mar = c(0,4,2,0))plot(sampleTree, main = \"Sample clustering to detect outliers\", sub=\"\", xlab=\"\", cex.lab = 2, cex.axis = 2, cex.main = 2)dev.off()#设置离群阈值####hlevel=280#绘制分割线####pdf(file = \"样品分割线_GEO.pdf\", width = 10, height = 10)par(cex = 0.5)par(mar = c(0,4,2,0))plot(sampleTree, main = \"Sample clustering to detect outliers\", sub=\"\", xlab=\"\", cex.lab = 2, cex.axis = 2, cex.main = 2)abline(h = hlevel, col = \"red\")#【h】是设置删除高度dev.off()#保留非离群样本####clust = cutreeStatic(sampleTree, cutHeight = hlevel, minSize = 10)##查看样本聚类信息####table(clust)##提取非离群数据####keepSamples = (clust==1)##保存非离群样本表达量####GEOdata_in=GEOdata[,keepSamples]###对剔除离群后的数据进行归一化####GEOdata_in=normalizeBetweenArrays(GEOdata_in)###对剔除后数据进行PCA分析####group_in=group[keepSamples,]dat_in=as.data.frame(t(GEOdata_in))dat.pca_in <- PCA(dat_in, graph = FALSE)pca_plot_in <- fviz_pca_ind(dat.pca_in,geom.ind = \"point\", col.ind = group_in$group, palette = randomColors[3:4], addEllipses = TRUE, legend.title = \"Groups\")ggsave(plot = pca_plot_in,filename =\"PCA_GEO_in.pdf\")#查看QC前后箱线图####pdf(file = \"box.pdf\",width = 15,height = 15)boxplot(GEOdata, names = NA, col = as.factor(group$group))legend(\"topright\", legend = unique(group$group), fill = as.factor(unique(group$group)), box.col = NA, bg = \"white\", inset = 0.01)dev.off()pdf(file = \"box_QC.pdf\",width = 15,height = 15)boxplot(GEOdata_in, names = NA, col = as.factor(group_in$group))legend(\"topright\", legend = unique(group_in$group), fill = as.factor(unique(group_in$group)), box.col = NA, bg = \"white\", inset = 0.01)dev.off()#导出数据####GEOdata_in<-as.data.frame(GEOdata_in)fwrite(GEOdata_in,\"GEO_out.txt\",quote = F,sep = \"\\t\",row.names = T)附xhhGEOQC.R代码xhhGEOQC<-function(GEOexp){qx <- as.numeric(quantile(GEOexp, c(0., 0.25, 0.5, 0.75, 0.99, 1.0), na.rm=T))LogC 100)|| (qx[6]-qx[1] > 50 && qx[2] > 0) || (qx[2]>0&&qx[2]1&&qx[4]<2)if(LogC){ message(\"需要取log2\")}else{message(\"无需取log2\") }}2.2 质控前PCA主成分分析和分组样品聚类结果
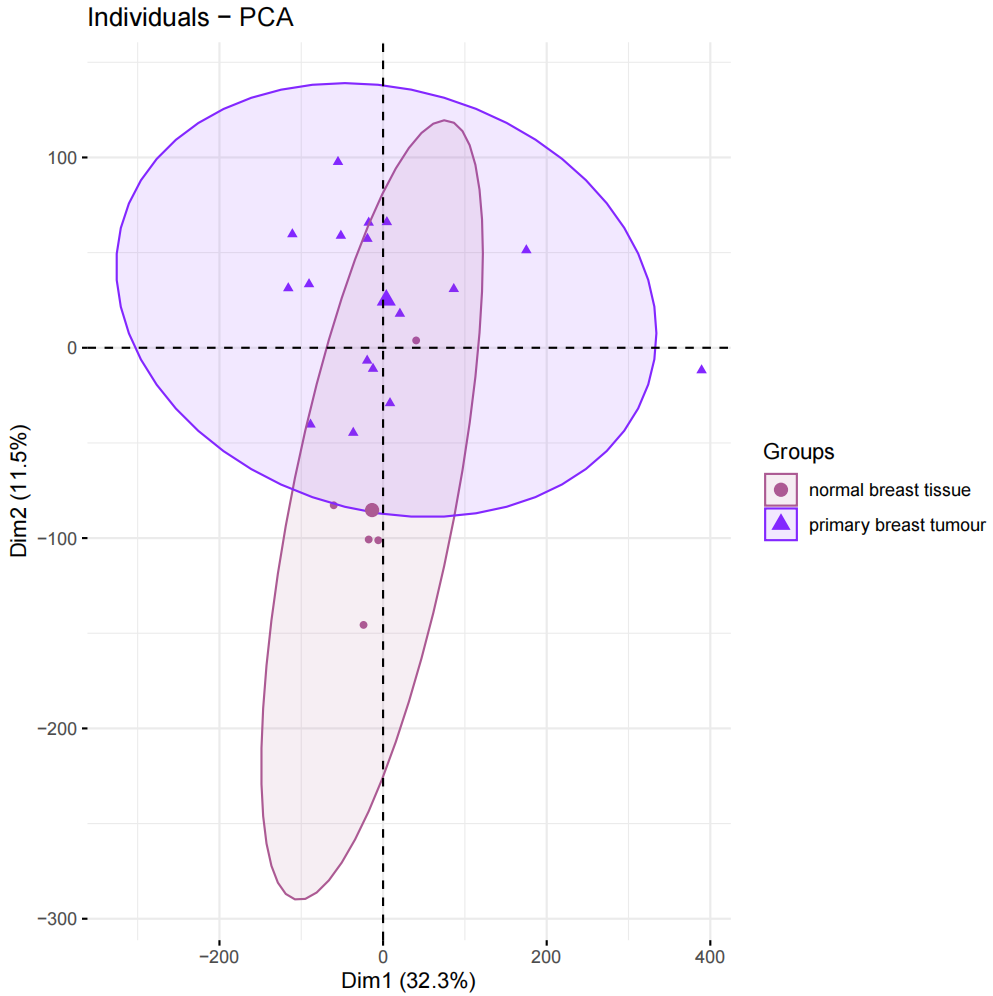

2.2.3 去除离群样本分割线
2.2.4 质控后PCA主成分分析和分组样品聚类结果
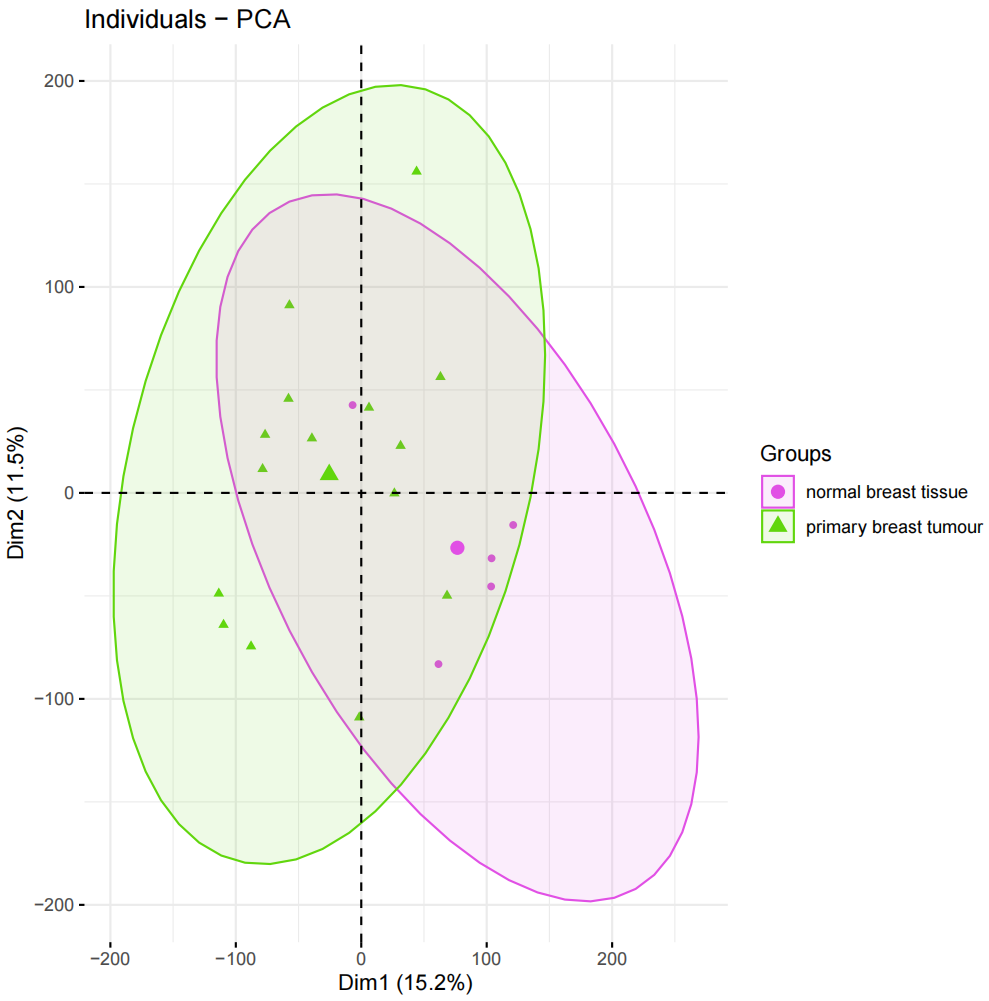
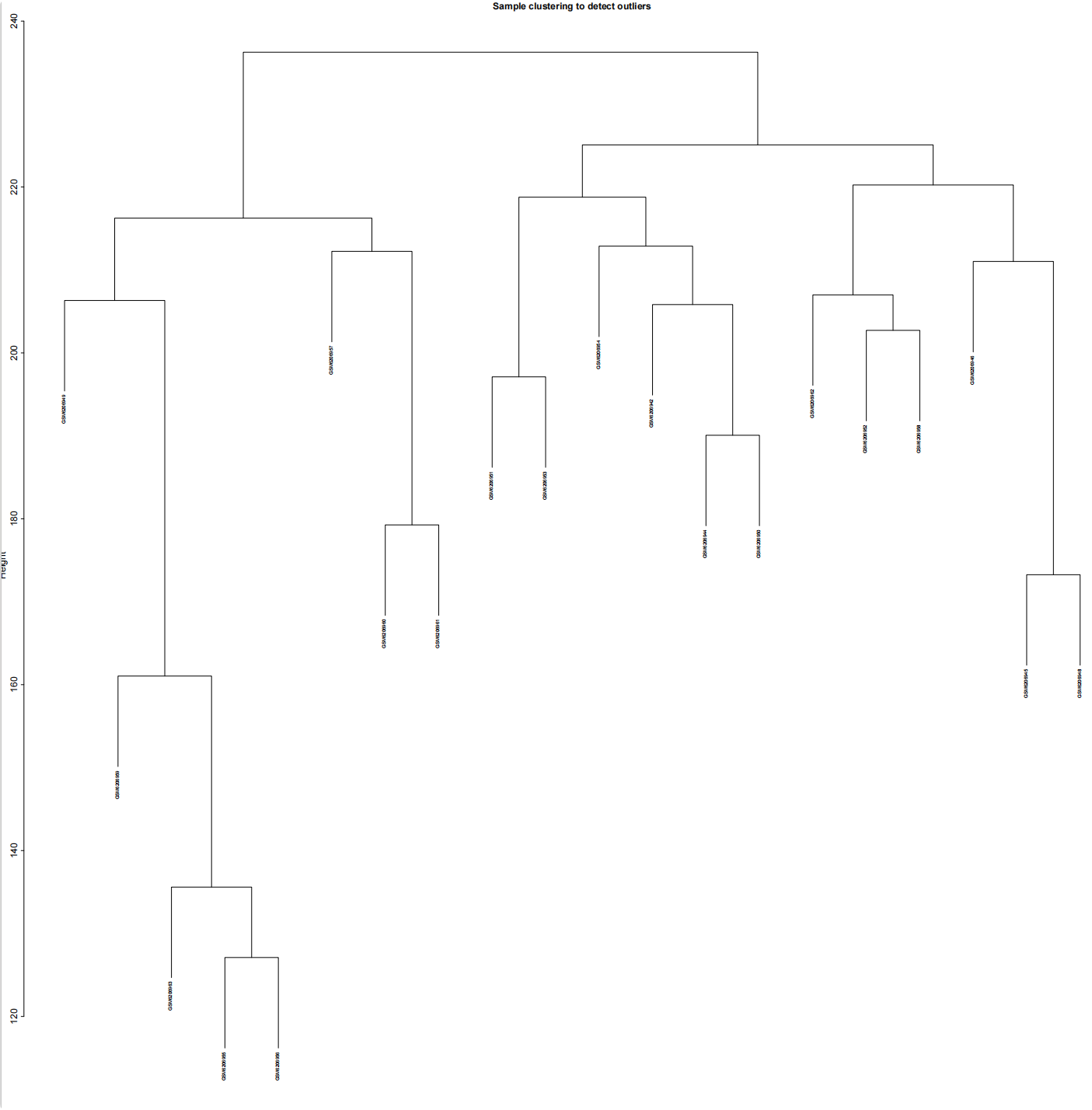
三、差异表达分析
3.1 DEseq2差异分析
使用limma包进行差异表达分析,设置阈值:
调整后p值(adj.P.Val) < 0.05·
绝对log2倍数变化(logFC) > 1
library(ggrepel)library(tidyverse)library(ggsignif) library(limma)library(ggplot2)library(ggpubr)library(ggthemes)library(beepr)library(gplots)library(parallel)library(future.apply)library(pheatmap)#读取表达数据####data<-fread(\"GEO_out.txt\",header = T,check.names = F,data.table = F)rownames(data)=data$V1data=data[,2:ncol(data)]#读取分组####groupdata<-fread(\"GEOpdata.txt\",header = T,check.names = F,data.table = F)group=data.frame(ID=groupdata$V1, group=groupdata$group)rownames(group)=group$ID#提取质控后的样本samesample<-intersect(rownames(group),colnames(data))group<-group[samesample,]data<-data[,samesample]#创建设计矩阵design<-data.frame(\"con\"=ifelse(group$group==\"normal breast tissue\",1,0), \"treat\"=ifelse(group$group==\"primary breast tumour\",1,0))#进行limma差异分析#######算方差####df.fit <- lmFit(data,design)df.matrix<- makeContrasts(treat - con ,levels=design)fit<- contrasts.fit(df.fit,df.matrix)##贝叶斯检验####fit2 <- eBayes(fit)##输出基因####allDEG1 = topTable(fit2,coef=1,n=Inf,adjust=\"BH\") allDEG1 = na.omit(allDEG1)##提取基因差异显著的差异矩阵####limma_diff = allDEG1[(allDEG1$adj.P.Val =1 | allDEG1$logFC<=(-1))),]#以校正后p=1为条件筛选差异基因limma_diff = limma_diff[order(limma_diff$logFC),]#以logFC值进行排序all_limma=allDEG1all_limma=all_limma[order(all_limma$adj.P.Val),]#以校正后p值进行排序fwrite(limma_diff,\"limma_diff.txt\",quote = F,sep = \"\\t\",row.names = T)#导出有差异的limma计算结果fwrite(all_limma,\"all_limma.txt\",quote = F,sep = \"\\t\",row.names = T)#导出所有limma计算结果limma_diff_exp=data[rownames(limma_diff),]#提取差异基因表达量fwrite(limma_diff_exp,\"limma_diff_exp.txt\",quote = F,sep = \"\\t\",row.names = T)#导出差异基因表达量3.2 结果可视化
3.2.1 基础火山图:展示差异基因的整体分布
#绘制基础火山图####all_limma$logp<--log10(all_limma$adj.P.Val) all_limma$Group=1 & all_limma$adj.P.Val<0.05),\"Up\",ifelse((all_limma$logFC<=-1 & all_limma$adj.P.Val<0.05),\"Down\",\"Not\"))limmaUP<-all_limma[all_limma$Group==\"Up\",]limmaUP<-limmaUP[order(limmaUP$adj.P.Val),]limmaDown<-all_limma[all_limma$Group==\"Down\",]limmaDown<-limmaDown[order(limmaDown$adj.P.Val),]plot<-ggscatter(all_limma, x=\"logFC\", y=\"logp\", color=\"Group\", palette=c(\"#2f5688\",\"#BBBBBB\",\"#CC0000\"), size=0.25, xlab=\"log2FoldChange\", ylab=\"-log10(Adjust P-value)\",)+ theme_base()+ geom_hline(yintercept=-log10(0.05),linetype=\"dashed\")+ geom_vline(xintercept=c(-1,1),linetype=\"dashed\")pdf(\"基础火山图.pdf\")print(plot)dev.off()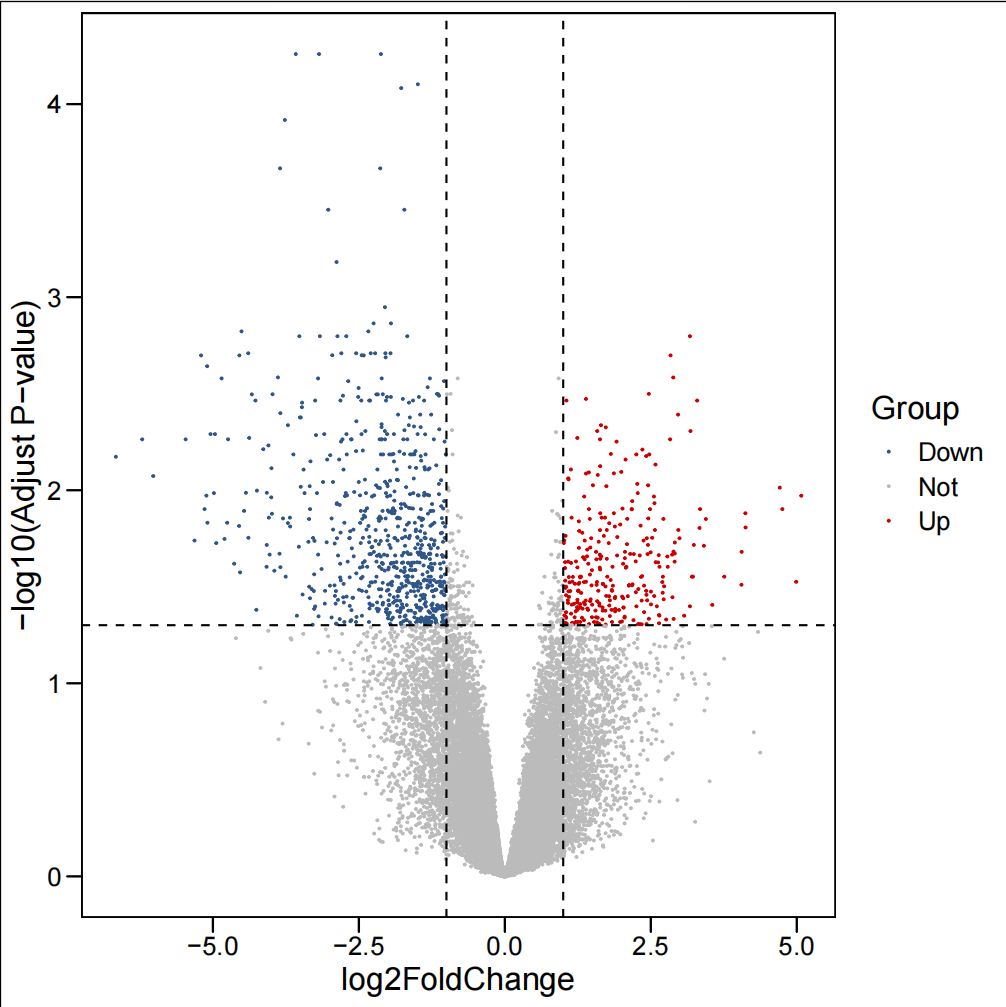
3.2.2 最显著差异基因火山图(5个)
#显著差异基因火山图####geneList0 <- c(rownames(limmaUP)[1:5],rownames(limmaDown)[1:5],rep(NA,(nrow(all_limma)-10)))geneList <-all_limma[geneList0,]plot1<-ggplot( all_limma, aes(x = logFC, y = -log10(adj.P.Val), color=Group)) + geom_point(alpha=0.5, size=0.25) + scale_colour_manual(values = c(\'blue\',\'gray\',\'red\'))+ geom_point(data=geneList,aes(x =logFC, y = -log10(adj.P.Val)),colour=\"yellow\",size=2)+ geom_vline(xintercept=c(-1,1),lty=3,col=\"black\",lwd=0.8) + geom_hline(yintercept = -log10(0.05),lty=3,col=\"black\",lwd=0.8) + labs(x=\"log2(fold change)\",y=\"-log10 (p-value)\")+ theme_bw()+ theme(panel.grid = element_blank())all_limma1<-all_limmaall_limma1$gene <- rownames(all_limma1)rownames(all_limma1) <- 1:nrow(all_limma1)geneList <- as.data.frame(geneList0)geneList[,2] <- geneListcolnames(geneList) <- c(\'gene\',\'label2\')c <-merge(all_limma1,geneList,by=\'gene\',all.x=TRUE)pdf(\"显著差异基因火山图.pdf\")plot1+ geom_label_repel(data = c, aes(x = logFC, y = -log10(adj.P.Val), label = label2), size = 2,color=\"black\", show.legend = FALSE)dev.off()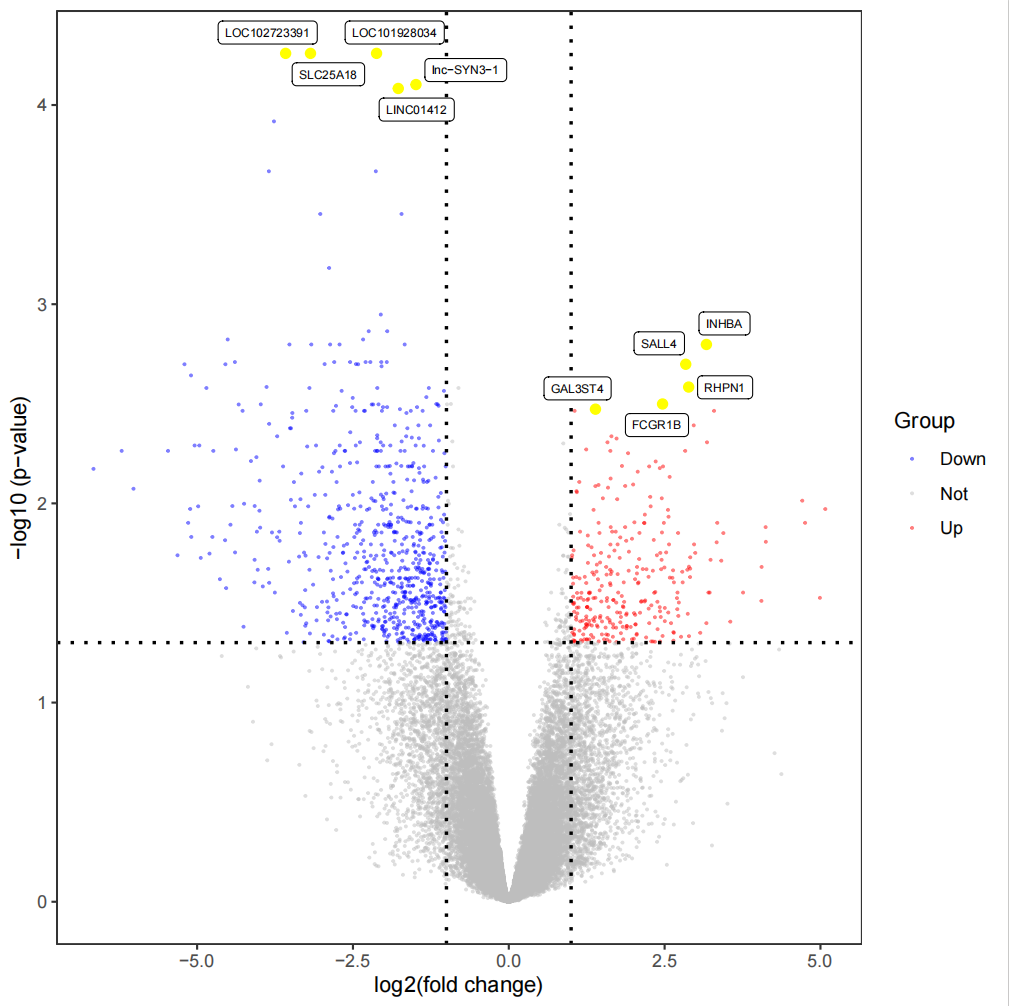
3.2.3 热图:展示上下调各50个差异基因的表达模式
#分组热图#####上调下调各取50个基因upgene<-rownames(limmaUP[order(limmaUP$logFC,decreasing = TRUE),])[1:50]downgene<-rownames(limmaDown[order(limmaDown$logFC,decreasing = TRUE),])[1:50]diffexp<-limma_diff_exp[c(upgene,downgene),]diffgroupmap<-group[order(group$group),]diffexp<-diffexp[,rownames(diffgroupmap)]diffgroupmap1<-data.frame(group=diffgroupmap$group)rownames(diffgroupmap1)<-rownames(diffgroupmap)pheatmaps<-pheatmap(diffexp, color=colorRampPalette(c(\"#3300CC\",\"white\",\"#CC0000\"))(50),border_color=NA, labels_row=NULL,show_rownames=T,show_colnames=F,annotation_col =diffgroupmap1,scale = \"row\",cluster_cols=F)pdf(\"分组差异热图.pdf\",width = 15,height = 15)print(pheatmaps)dev.off()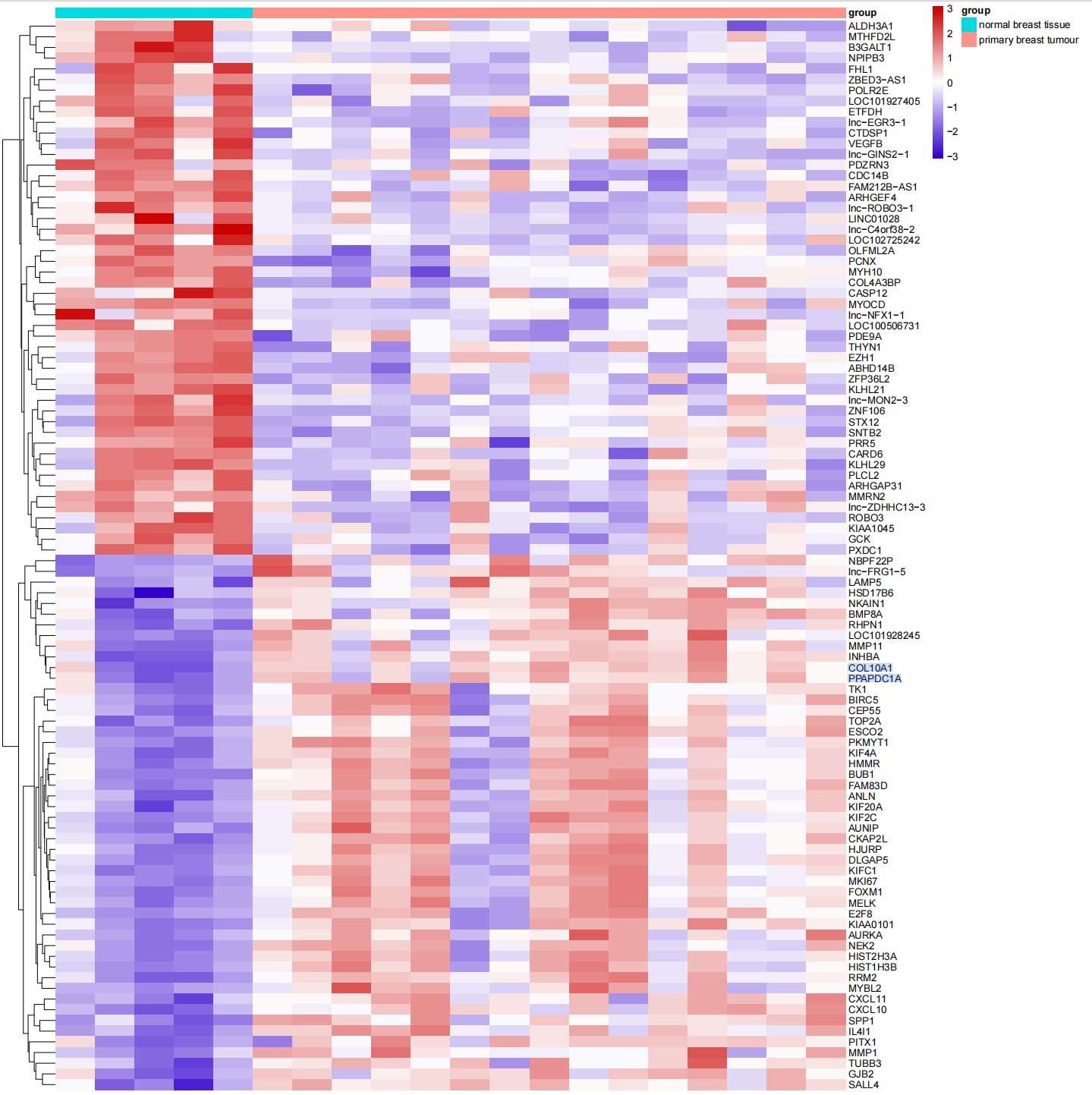
四、GO、KEGG富集分析
Gene Ontology(GO)分析揭示了差异基因参与的生物学过程、分子功能和细胞组分。
KEGG通路分析显示差异基因显著富集于关键通路
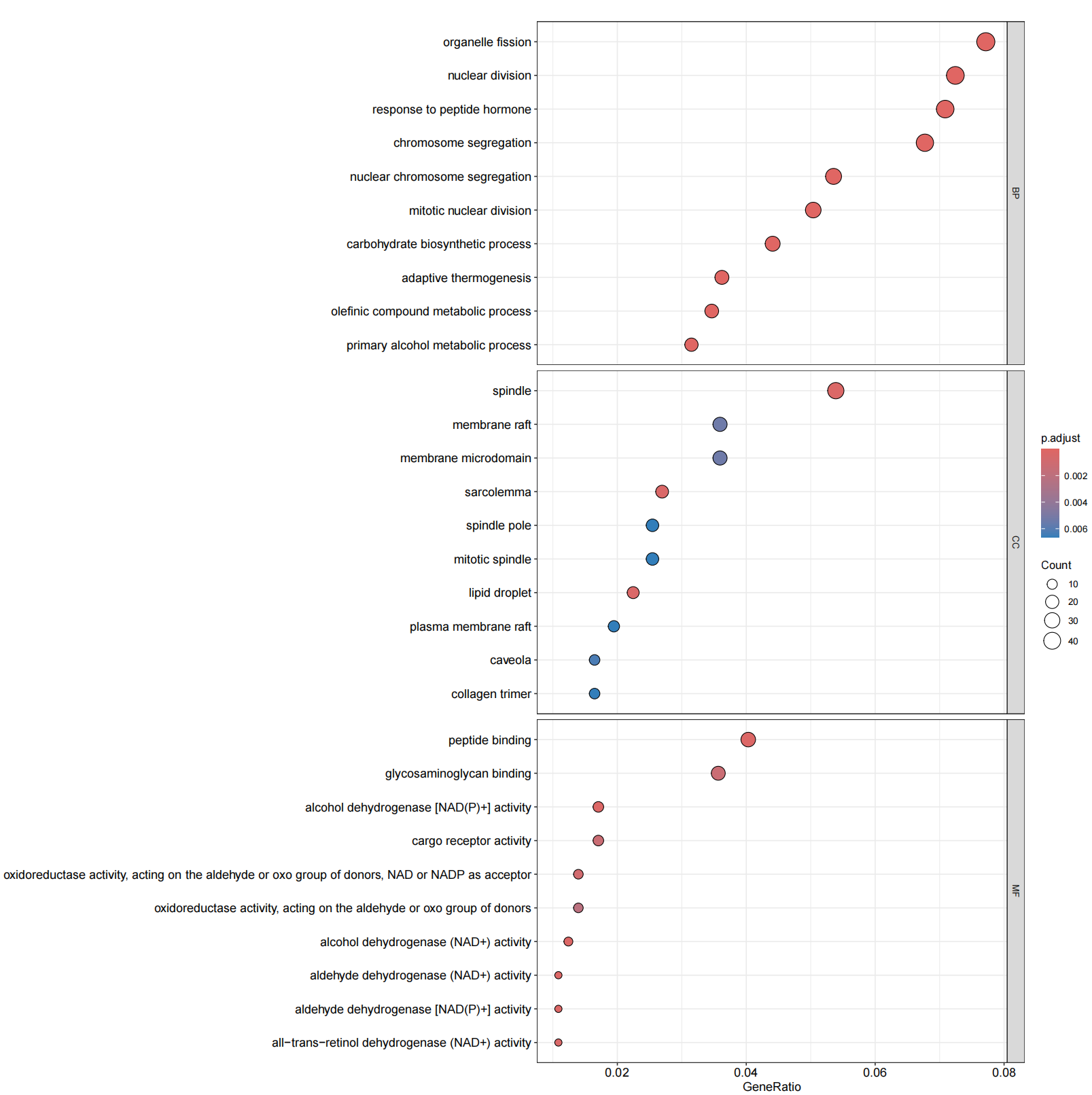
library(clusterProfiler)library(org.Hs.eg.db)library(enrichplot)library(ggplot2)library(org.Hs.eg.db) library(GOplot)library(data.table)library(doParallel)library(tidyverse)#转换IDrt=fread(\"limma_diff.txt\",sep=\"\\t\",check.names=F,header=T,data.table = F)rt=na.omit(rt)genes=rt$V1entrezIDs <- mget(genes, org.Hs.egSYMBOL2EG, ifnotfound=NA) #找出基因对应的identrezIDs <- as.character(entrezIDs)out=cbind(rt,entrezID=entrezIDs)out=out[out[,\"entrezID\"]!=\"NA\",]out$entrezID=as.numeric(out$entrezID)out=na.omit(out)gene=as.numeric(out$entrezID)gene=as.data.frame(gene)gene=na.omit(gene)gene=gene$gene#GO分析#######GO富集分析####kk <- enrichGO(gene = gene,OrgDb = org.Hs.eg.db, pvalueCutoff =0.05, qvalueCutoff = 0.05,ont=\"all\",readable =T)write.table(kk,file=\"GO.txt\",sep=\"\\t\",quote=F,row.names = F) ##可视化#######条形图####pdf(file=\"GO-barplot.pdf\",width = 10,height = 15)barplot(kk, drop = TRUE, showCategory =10,label_format=100,split=\"ONTOLOGY\") + facet_grid(ONTOLOGY~., scale=\'free\')dev.off()###气泡图####pdf(file=\"GO-bubble.pdf\",width = 10,height = 15)dotplot(kk,showCategory = 10,label_format=100,split=\"ONTOLOGY\") + facet_grid(ONTOLOGY~., scale=\'free\')dev.off()#KEGG####rt=fread(\"limma_diff.txt\",sep=\"\\t\",check.names=F,header=T) #读取文件rt=na.omit(rt)genes=rt$V1entrezIDs <- mget(genes, org.Hs.egSYMBOL2EG, ifnotfound=NA) #找出基因对应的identrezIDs <- as.character(entrezIDs)out=cbind(rt,entrezID=entrezIDs)write.table(out,file=\"KEGG-id.txt\",sep=\"\\t\",quote=F,row.names=F) #输出结果rt=read.table(\"KEGG-id.txt\",sep=\"\\t\",header=T,check.names=F) rt=rt[rt[,\"entrezID\"]!=\"NA\",] rt$entrezID=as.numeric(rt$entrezID)rt=na.omit(rt)gene=rt$entrezIDgene=as.data.frame(gene)gene=na.omit(gene)gene=gene$gene##kegg分析####kk <- enrichKEGG(gene = gene, keyType = \"kegg\", organism = \"hsa\", pvalueCutoff =0.05, qvalueCutoff =0.05, pAdjustMethod = \"fdr\") write.table(kk,file=\"KEGG.txt\",sep=\"\\t\",quote=F,row.names = F) ##可视化#######条形图####pdf(file=\"KEGG-barplot.pdf\",width = 10,height = 13)barplot(kk, drop = TRUE, showCategory = 15,label_format=100)dev.off()###气泡图####pdf(file=\"KEGG-bubble.pdf\",width = 10,height = 13)dotplot(kk, showCategory = 15,label_format=100)dev.off()GO富集条形图
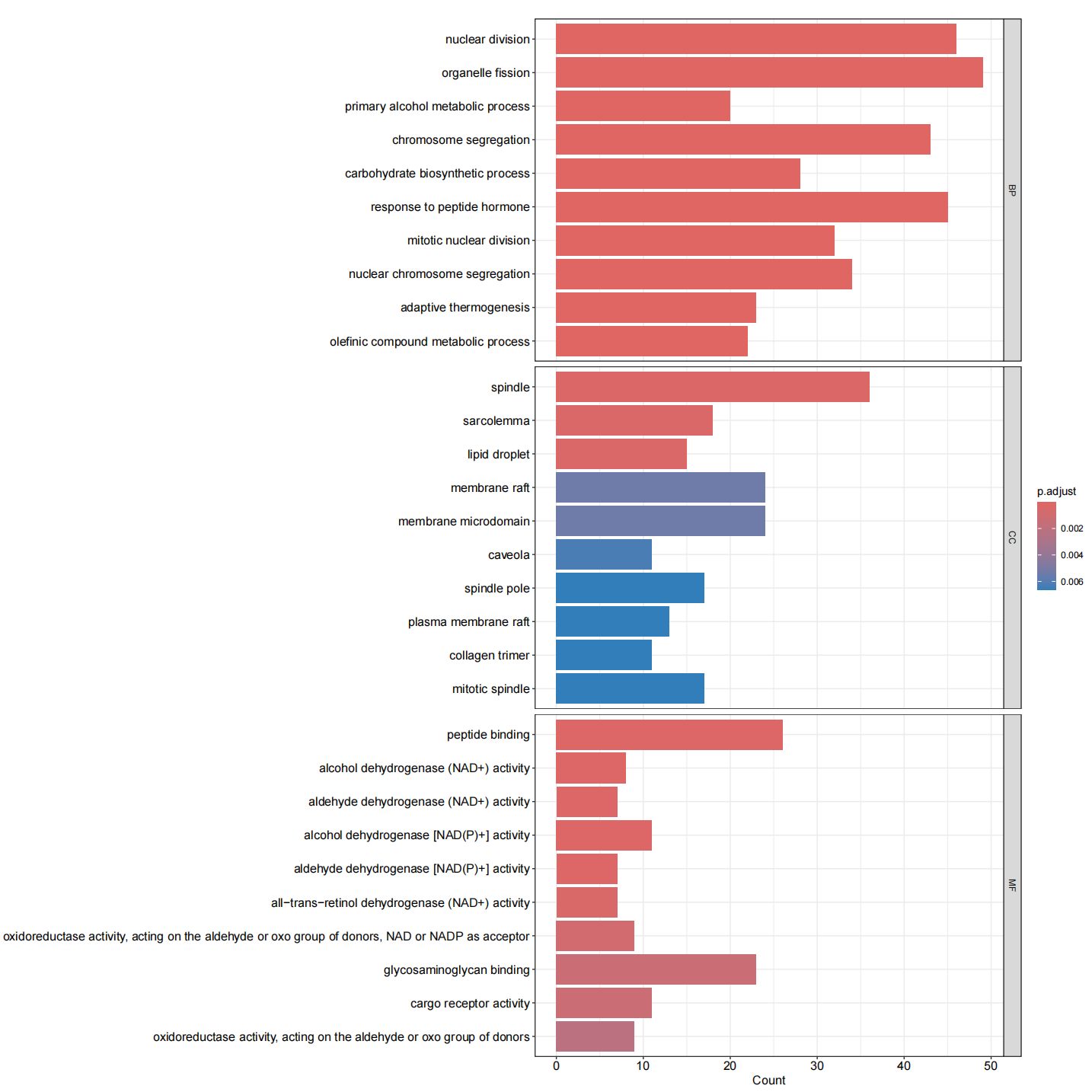
GO富集气泡图
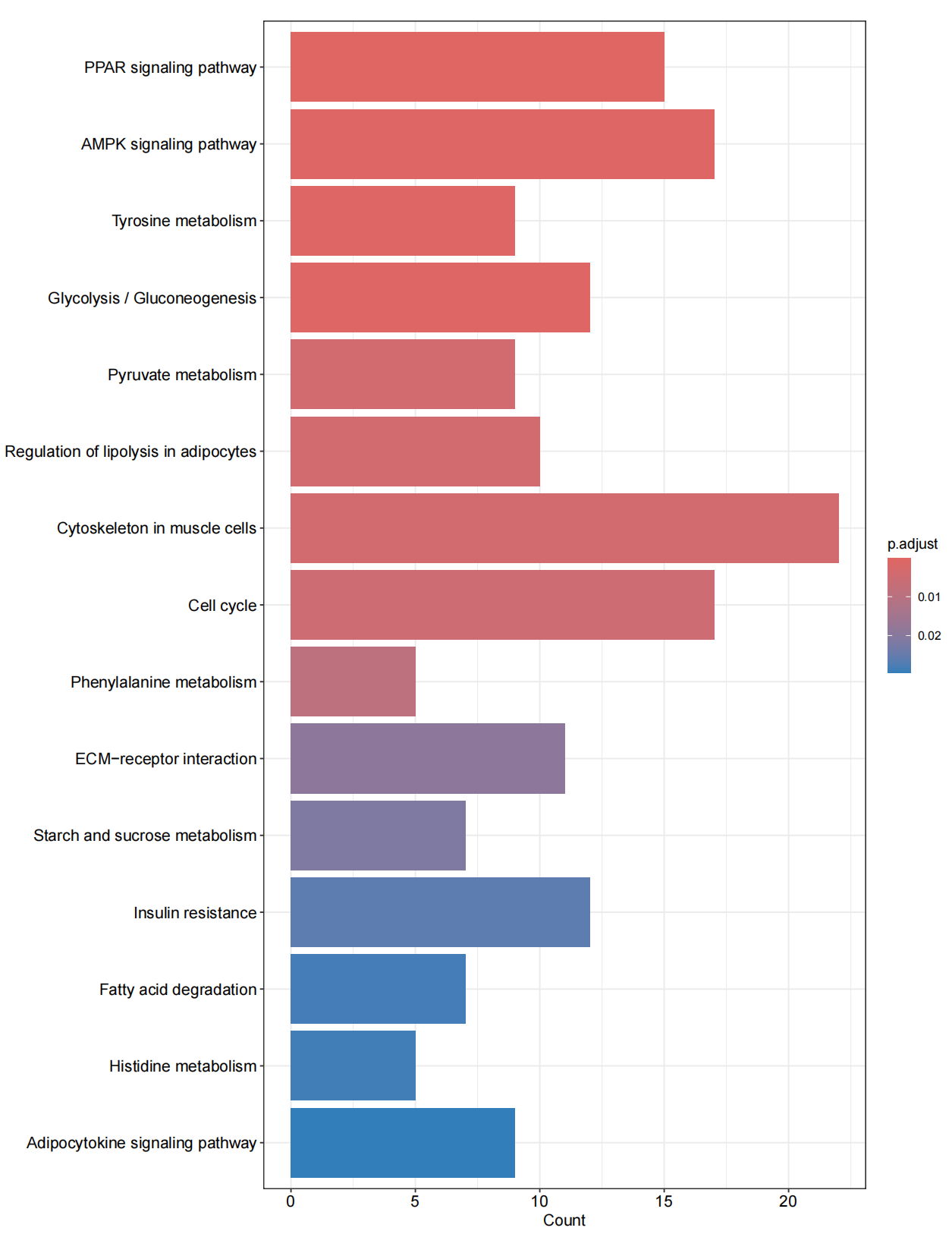
KEGG富集条形图
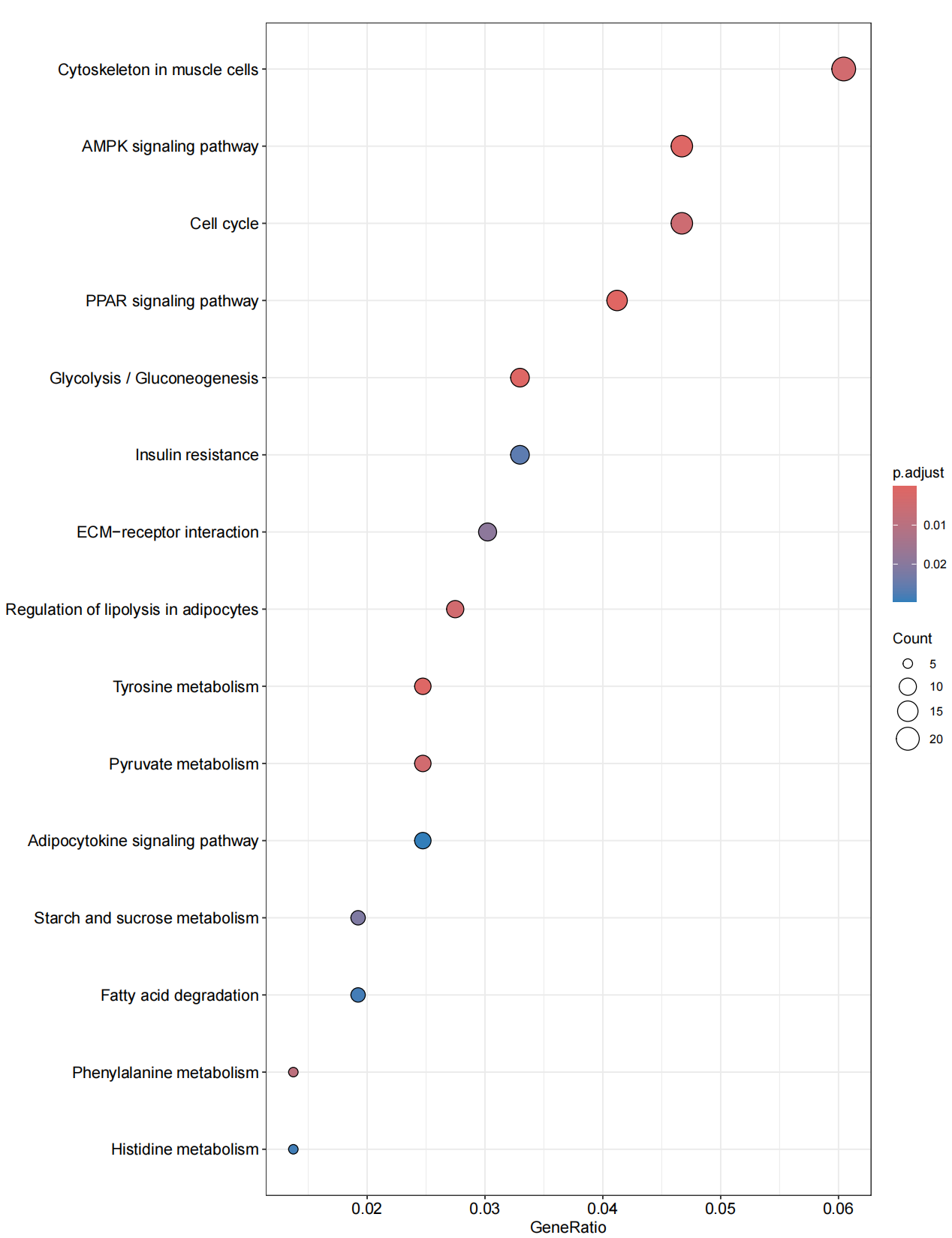
KEGG富集气泡图
五、单基因GSEA富集
5.1 GSEA富集的目的是观察我们研究的基因通过哪些功能或通路来影响疾病的发生。
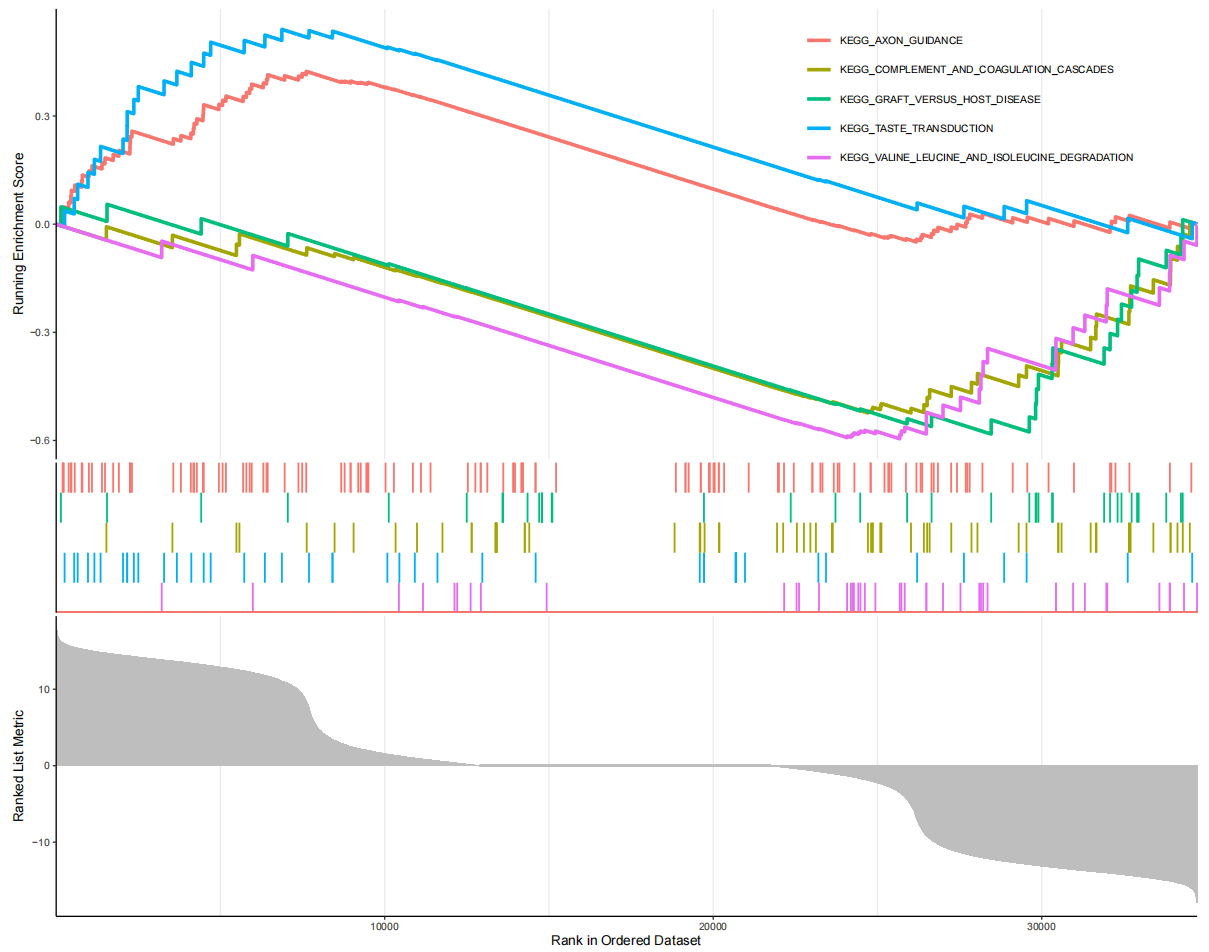
#加载R包####library(limma)library(tidyverse)library(org.Hs.eg.db)library(clusterProfiler)library(enrichplot)library(data.table)library(doParallel)#读取表达数据####data=fread(\"GEO_out.txt\",sep = \"\\t\",header = T,check.names = F,data.table = F)rownames(data)=data$V1data=data[,2:ncol(data)]#读取分组####groupdata<-fread(\"GEOpdata.txt\",header = T,check.names = F,data.table = F)rownames(groupdata)<-groupdata$V1samesample<-intersect(colnames(data),rownames(groupdata))data<-data[,samesample]groupdata<-groupdata[samesample,]#提取肿瘤数据####Tumordata<-data[,rownames(groupdata[groupdata$group==\"primary breast tumour\",])]#高低表达组比较,得到logFC#高data低表达组比较,得到logFCgene=\"INHBA\"dataL=data[,as.numeric(Tumordata[gene,])median(as.numeric(Tumordata[gene,])),drop=F]meanL=rowMeans(dataL)meanH=rowMeans(dataH)meanL[meanL<0.00001]=0.00001meanH[meanH=termNum){showTerm=row.names(kkTab)[1:termNum]gseaplot=gseaplot2(kk, showTerm, base_size=8, title=\"\")pdf(file=\"GSEA-GO.pdf\", width=10, height=8)print(gseaplot)dev.off()}else{showTerm=row.names(kkTab)gseaplot=gseaplot2(kk, showTerm, base_size=8, title=\"\")pdf(file=\"GSEA-GO.pdf\", width=10, height=8)print(gseaplot)dev.off()}#####KEGG######读入基因集文件gmt=read.gmt(\"c2.cp.kegg_legacy.v2024.1.Hs.symbols.gmt\")#富集分析kk=GSEA(logFC, TERM2GENE=gmt, pvalueCutoff = 1)kkTab=as.data.frame(kk)write.table(kkTab,file=\"GSEA.result-KEGG.txt\",sep=\"\\t\",quote=F,row.names = F)#输出富集的图形termNum=5 #展示前5个通路if(nrow(kkTab)>=termNum){ showTerm=row.names(kkTab)[1:termNum] gseaplot=gseaplot2(kk, showTerm, base_size=8, title=\"\") pdf(file=\"GSEA-KEGG.pdf\", width=10, height=8) print(gseaplot) dev.off()}else{ showTerm=row.names(kkTab) gseaplot=gseaplot2(kk, showTerm, base_size=8, title=\"\") pdf(file=\"GSEA-KEGG.pdf\", width=10, height=8) print(gseaplot) dev.off()}5.2 单基因富集结果:最多富集五个通路,不足五个按实际通路个数展示。左边灰色是和我们目标基因是正相关的基因,右边灰色是负相关基因,通路的峰值在左边表示与目标基因表达成正相关,目标基因表达越高,这些通路越活跃。
GSEA-GO富集
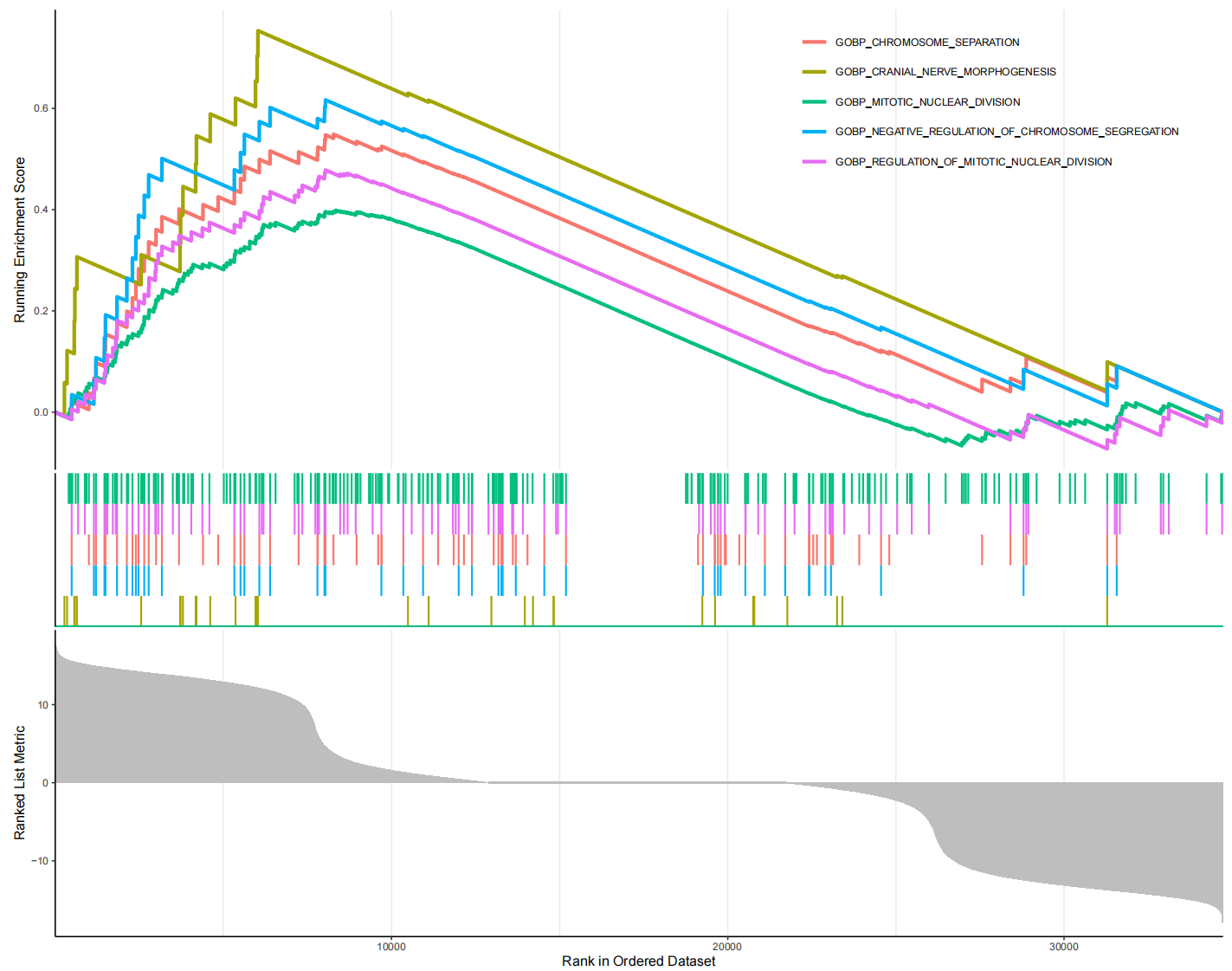
GSEA-KEGG富集
六、免疫浸润分析
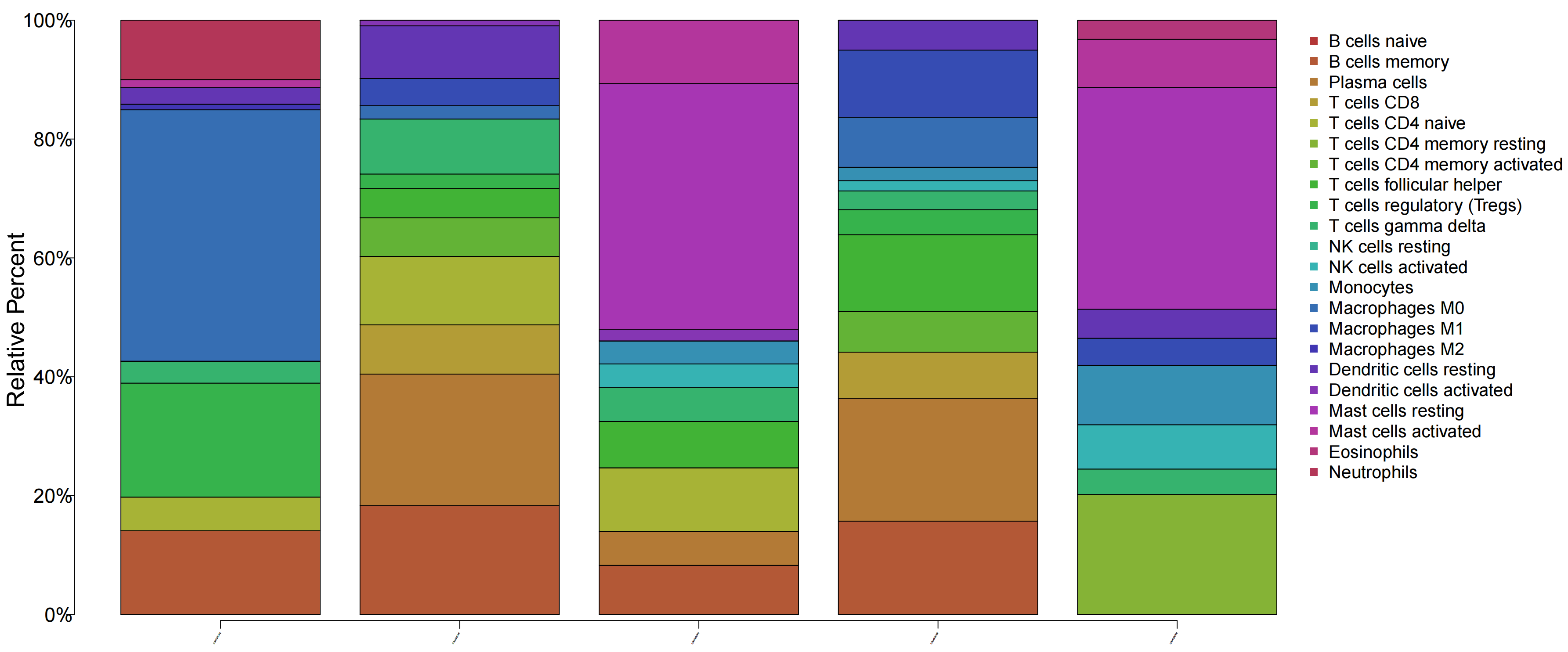
6.1 免疫浸润分析(Immune Infiltration Analysis)的主要目的是评估肿瘤或疾病微环境中免疫细胞的组成、丰度、功能状态,可以了解样本中不同免疫细胞的构成比例,以及不同细胞类型的相对丰度。
library(tcltk)library(limma)library(reshape2)library(ggpubr)library(vioplot)library(ggExtra)library(data.table)library(tidyverse)library(doParallel)#读取数据####data=fread(\"GEO_out.txt\",header = T,check.names = F,data.table = F)rownames(data)=data$V1data0=data[,2:ncol(data)] out=rbind(ID=colnames(data0), data0)fwrite(out,file=\"uniq.symbol.txt\",sep=\"\\t\",quote=F,col.names=F,row.names = T)#运行主代码,得到免疫细胞浸润的结果source(\"xxdimmune.R\",encoding =\"utf-8\" )results=CIBERSORT(\"LM22.txt\", \"uniq.symbol.txt\", perm=1000, QN=TRUE)immune=read.table(\"CIBERSORT-Results.txt\",sep=\"\\t\",header=T,row.names=1,check.names=F)immune=immune[immune[,\"P-value\"]=median(rt$gene),\"High\",\"Low\")data=rt[,-(ncol(rt)-1)]data=melt(data,id.vars=c(\"gene\"))colnames(data)=c(\"gene\", \"Immune\", \"Expression\")#绘制箱线图group=levels(factor(data$gene))data$gene=factor(data$gene, levels=c(\"Low\",\"High\"))bioCol=c(\"#223D6C\",\"#FFD121\")bioCol=bioCol[1:length(group)]boxplot=ggboxplot(data, x=\"Immune\", y=\"Expression\", fill=\"gene\", xlab=\"\", ylab=\"Fraction\", legend.title=gene, width=0.8, palette=bioCol)+ rotate_x_text(50)+ stat_compare_means(aes(group=gene),symnum.args=list(cutpoints=c(0, 0.001, 0.01, 0.05, 1), symbols=c(\"***\", \"**\", \"*\", \"\")), label=\"p.signif\")#输出图片pdf(file=\"immune_diff.pdf\", width=7, height=6)print(boxplot)dev.off()6.2 22种免疫细胞在p<0.05样本中的占比分布情况
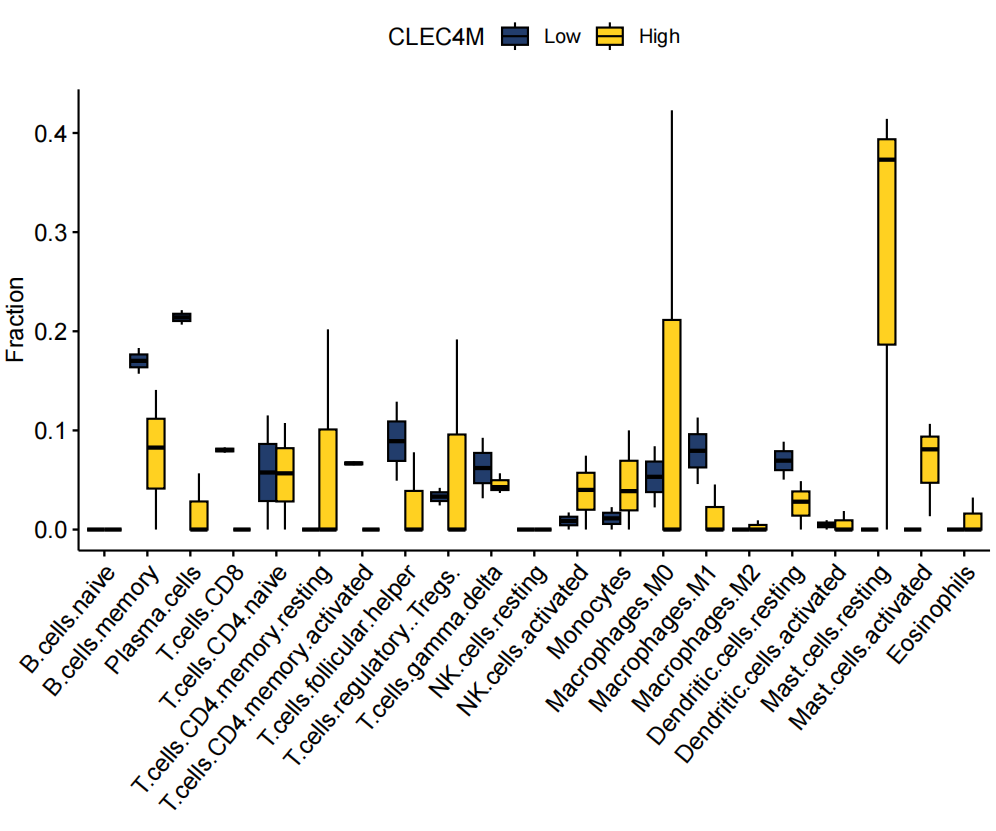
6.3 单个基因在每个细胞的表达水平
七、WGCNA分析
7.1 WGCNA分析目的是找寻协同表达的基因模块,并探索基因网络与关注的表型之间的关系,以及网络中的核心基因
library(WGCNA)library(limma)library(tidyverse)library(tcltk)library(edgeR)library(data.table)library(doParallel)library(Rcpp)library(RcppArmadillo)allowWGCNAThreads(nThreads=6)enableWGCNAThreads(nThreads=6)cl <- makeCluster(6) # 根据实际情况选择较小的核心数registerDoParallel(cl)#多线程工作setDTthreads(threads =detectCores())#读取表达数据####data=fread(\"GEO_out.txt\",sep = \"\\t\",header = T,check.names = F,data.table = F)rownames(data)=data$V1data=data[,2:ncol(data)]dataWGCNA<-t(data)#样品分群聚类###sampleTree = hclust(dist(dataWGCNA), method = \"average\")#【将每个相似的样品进行聚类】pdf(file = \"1_聚类图.pdf\", width = 10, height = 10)#【绘制聚类图】par(cex = 0.5)par(mar = c(0,4,2,0))plot(sampleTree, main = \"Sample clustering to detect outliers\", sub=\"\", xlab=\"\", cex.lab = 2, cex.axis = 2, cex.main = 2)dev.off()hlevel=280#【根据聚类图,查看是否有离群值,并设置删除高度】###【删除离群值】pdf(file = \"1_聚类图切割线.pdf\", width = 10, height = 10)par(cex = 0.5)par(mar = c(0,4,2,0))plot(sampleTree, main = \"Sample clustering to detect outliers\", sub=\"\", xlab=\"\", cex.lab = 2, cex.axis = 2, cex.main = 2)abline(h = hlevel, col = \"red\")#【h】是设置删除高度dev.off()###【保留非离群样本】clust = cutreeStatic(sampleTree, cutHeight = hlevel, minSize = 10)table(clust)keepSamples = (clust==1)#【提取非离群数据】dataWGCNA = dataWGCNA[keepSamples, ] #【构建非离群矩阵】#读取免疫浸润结果####traitData = fread(\"CIBERSORT-Results.txt\",header = T,check.names = F,data.table = F)rownames(traitData) 1){ pdfFile=paste(\"9_\", trait, \"_\", module,\".pdf\",sep=\"\") outPdf=paste(picDir,pdfFile,sep=\"\\\\\") pdf(file=outPdf,width=7,height=7) par(mfrow = c(1,1)) verboseScatterplot(abs(geneModuleMembership[moduleGenes, column]), abs(geneTraitSignificance[moduleGenes, traitColumn]), xlab = paste(\"Module Membership in\", module, \"module\"), ylab = paste(\"Gene significance for \",trait), main = paste(\"Module membership vs. gene significance\\n\"), cex.main = 1.2, cex.lab = 1.2, cex.axis = 1.2, col = module) dev.off() } }}stopCluster(cl)#提取表达量#####rt1=read.table(\"module_turquoise.txt\",sep = \"\\t\")samegene=intersect(rt1$V1, rownames(data))GenesExp=data[samegene,,drop=F]fwrite(file = \"WGCNAgene_exp.txt\", GenesExp, sep = \"\\t\", quote = F, row.names = T)7.2
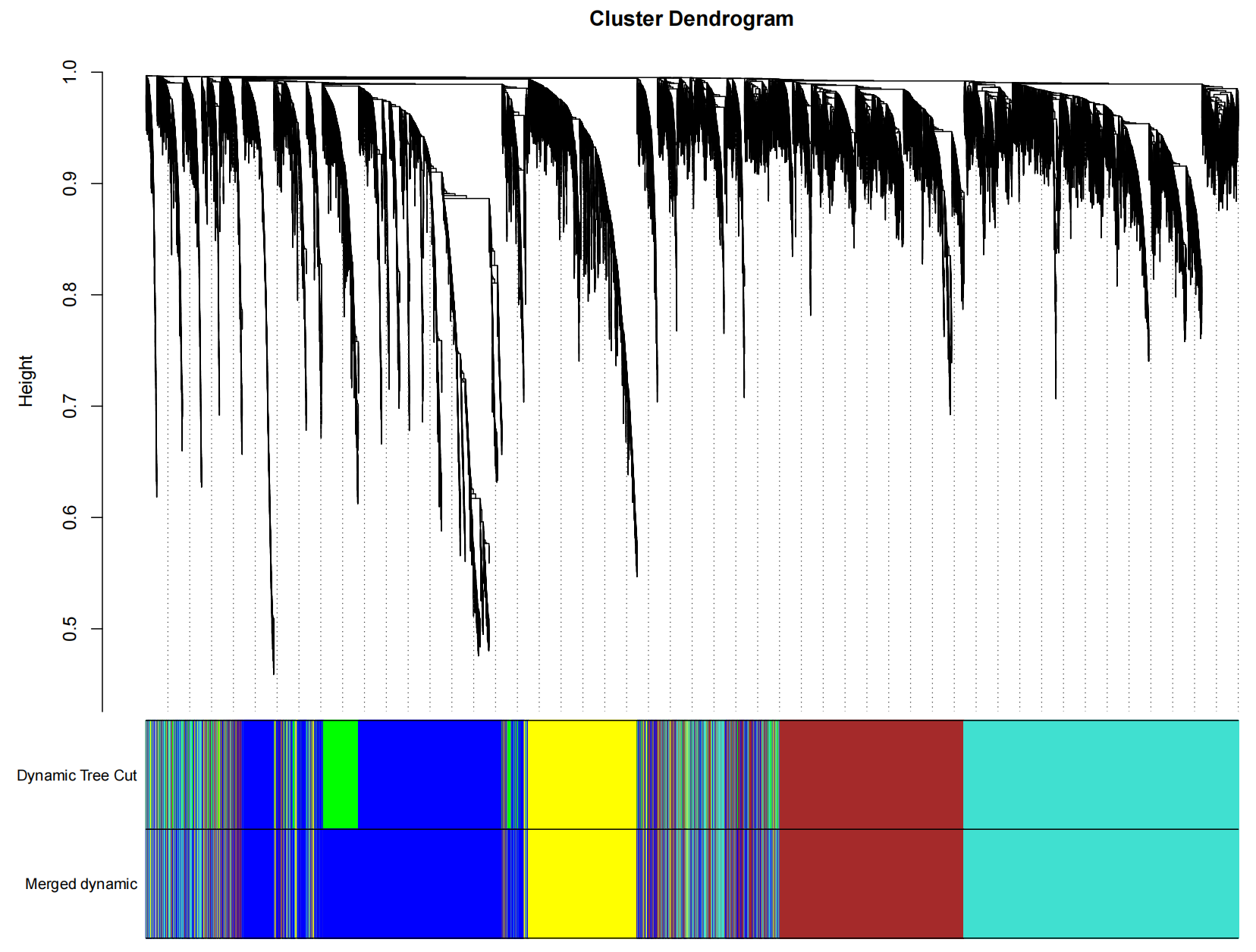
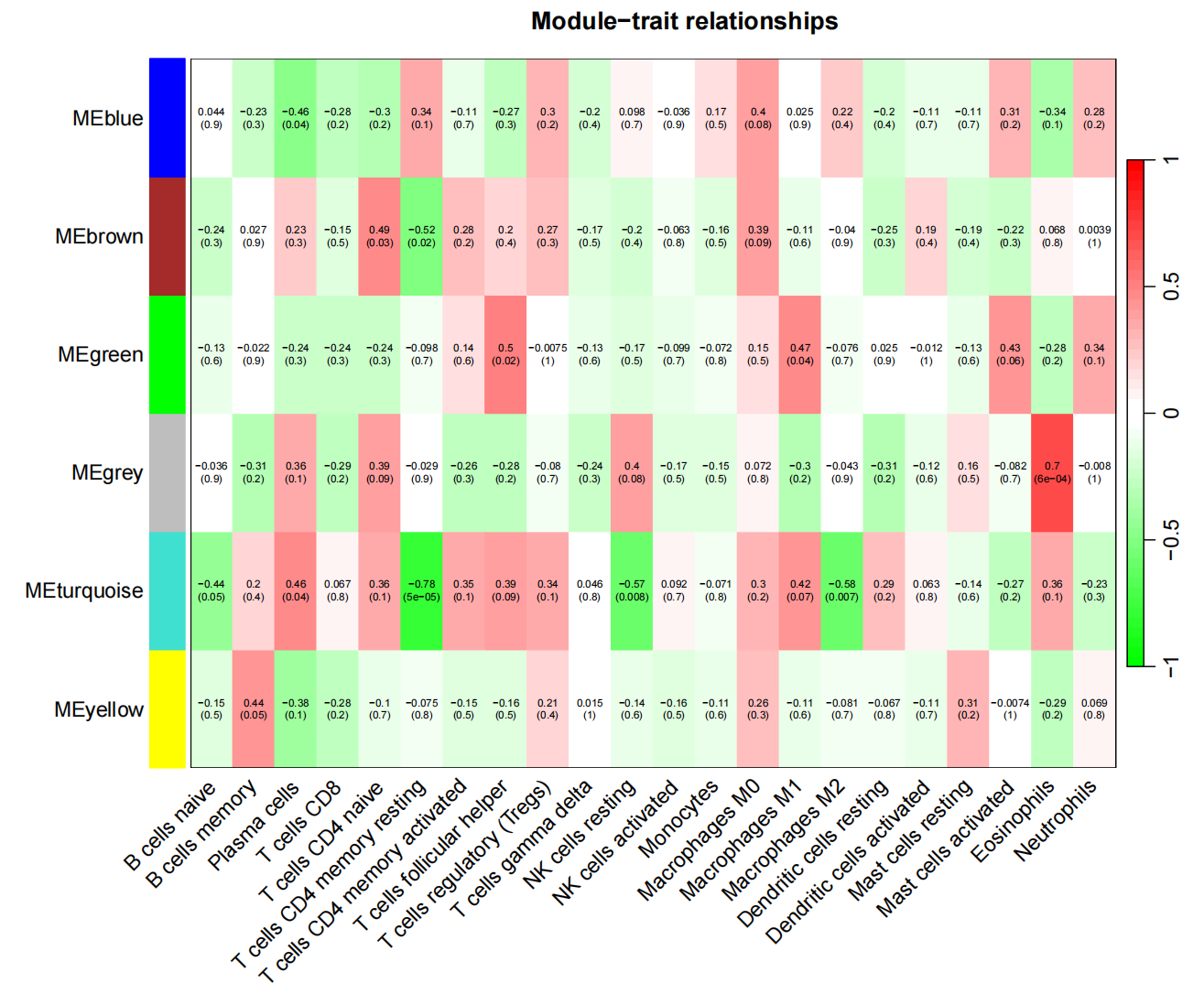
7.3 与表型(免疫细胞)的相关性结果,最显著相关的是MEturquoise板块的第一个绿色小板块(相关性-0.78,p=5e−05)
八、机器学习(lasso)筛选hub基因
根据WGCNA确定的与免疫细胞最相关的板块基因来筛选hub基因作为疾病标志物
library(data.table)library(ggrepel)library(tidyverse)library(ggsignif) library(limma)library(ggplot2)library(ggpubr)library(ggthemes)library(beepr)library(gplots)library(parallel)library(future.apply)library(pheatmap)library(glmnet) setDTthreads(threads =detectCores())#读取表达数据####data<-fread(\"WGCNAgene_exp.txt\",header = T,check.names = F,data.table = F)rownames(data)=data$V1data=data[,2:ncol(data)]#读取分组####groupdata<-fread(\"GEOpdata.txt\",header = T,check.names = F,data.table = F)group=data.frame(ID=groupdata$V1, group=groupdata$group)rownames(group)=group$IDsamesample<-intersect(rownames(group),colnames(data))group<-group[samesample,]data<-data[,samesample]design<-data.frame(\"con\"=ifelse(group$group==\"normal breast tissue\",1,0), \"treat\"=ifelse(group$group==\"primary breast tumour\",1,0))#进行limma差异分析#######算方差####df.fit <- lmFit(data,design)df.matrix<- makeContrasts(treat - con ,levels=design)fit<- contrasts.fit(df.fit,df.matrix)##贝叶斯检验####fit2 <- eBayes(fit)##输出基因####allDEG1 = topTable(fit2,coef=1,n=Inf,adjust=\"BH\") allDEG1 = na.omit(allDEG1)##提取基因差异显著的差异矩阵####limma_diff = allDEG1[(allDEG1$adj.P.Val =1 | allDEG1$logFC<=(-1))),]#以校正后p=1为条件筛选差异基因#进行lasso分析####set.seed(123)x=t(data)y=group$groupfit=glmnet(x, y, family = \"binomial\", alpha=1)cvfit=cv.glmnet(x, y, family=\"binomial\", alpha=1,type.measure=\'deviance\',nfolds = 10)coef=coef(fit, s = cvfit$lambda.min)index=which(coef != 0)lassoGene=row.names(coef)[index]lassoGenes=lassoGene[-1]#输出基因####geneouts<-data.frame(gene=lassoGenes)fwrite(geneouts,\"geneouts.txt\",,quote = F,sep = \"\\t\",row.names = T)

