(超详细)零基础RNA-Seq分析:表达矩阵的获取(Window系统下)_rnaseq fastq linux
参考文章:RNA-Seq:从fastq到表达矩阵 - 简书
目录
一、分析环境搭建
1. 在Windows系统下安装Linux子系统
2. Linux-conda环境安装
2.1 软件准备
2.2 镜像源设置
二、测序数据的准备
1. 从SRA数据库下载数据
2. 公司返回的测序下机数据
2.1 单端测序(Single-read)
2.2 双端测序(Paired-End)
三、对数据进行质控和过滤
1. fastp安装
2. 质控和过滤
四、参考基因组准备
1. 文件下载
2. 文件准备
五、比对到参考基因组
1. 构建索引
2. 使用Hisat2进行比对
3. 比对结果压缩、排序及构建索引
六、计算表达量
1. 工具包下载
2. 表达定量和标准化
3. 合并文件
4. 生成矩阵
一、分析环境搭建
1. 在Windows系统下安装Linux子系统
参考教程:Windows10安装Linux子系统Ubuntu 20.04LTS,轻松使用生信软件,效率秒杀虚拟机
具体流程如下:
(1)更新系统,确保Win10系统版本>1606,建议为最新版,提高系统兼容性和安全性:开始 —— 设置 —— 更新和安装。
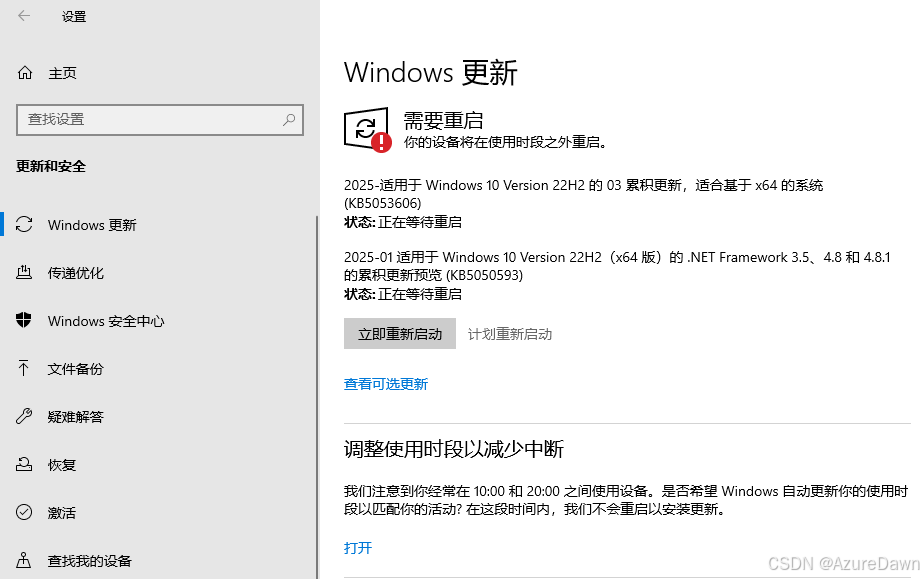
上图显示系统需要更新。“立即重新启动”可使已经安装更新生效。“下载和安装”可以继续安装更新。可能要反复下载更新重启几次。
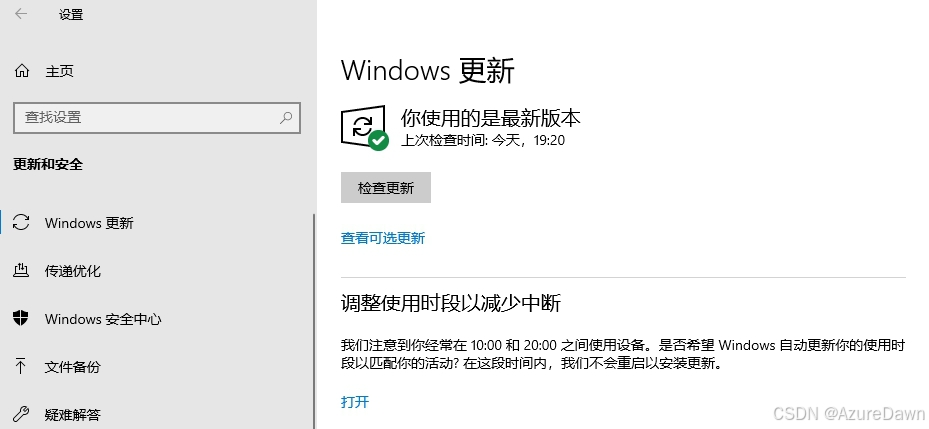
显示“你使用的是最新版本”,系统为最新版。
(2)启动开发人员模式
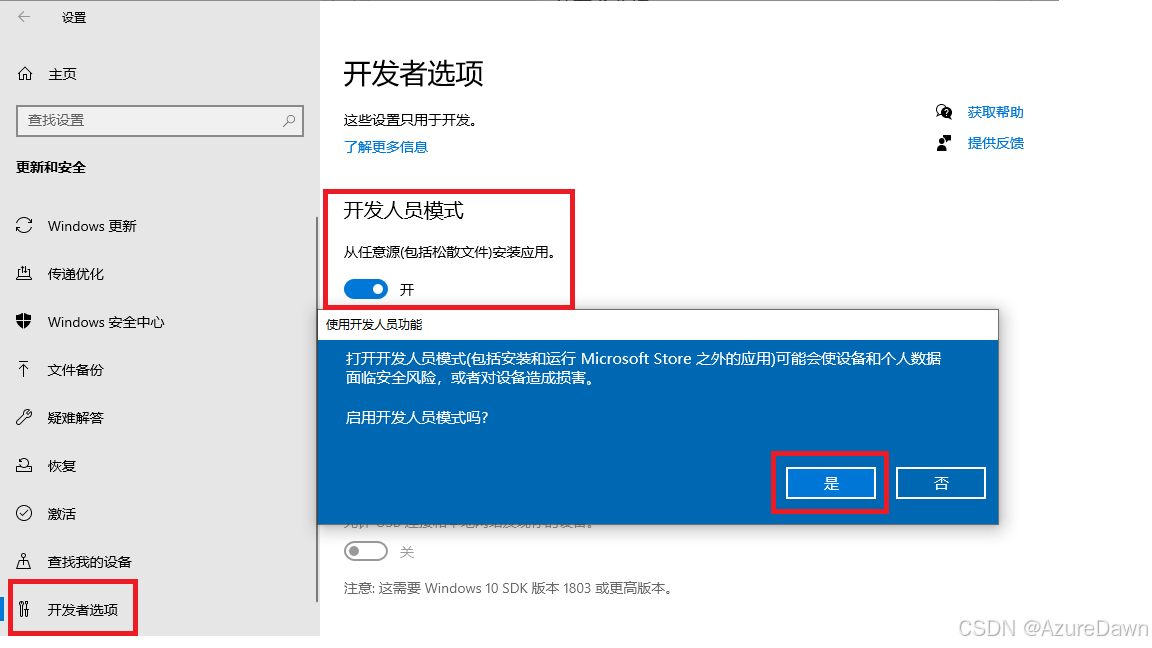
同一个页面,左侧选择“开发者选项” —— 切换至“开发人员模式”,点击“是”确认,会自动安装开发人员模式程序包,并启动桌面远程工具等。
(3)启动适用于Linux的Windows子系统
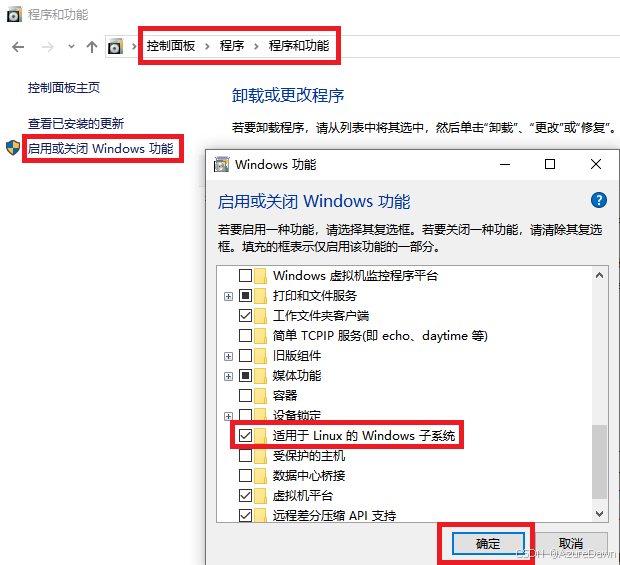
Win10开始菜单旁“搜索”按钮,查找“控制面板”并打开,选择”程序” —— “程序与功能”子页面,点击“启用或关闭Windows功能”,托动滚动条至最低部,勾远“适用于Linux的Windows子系统” ,再点击“确定”。
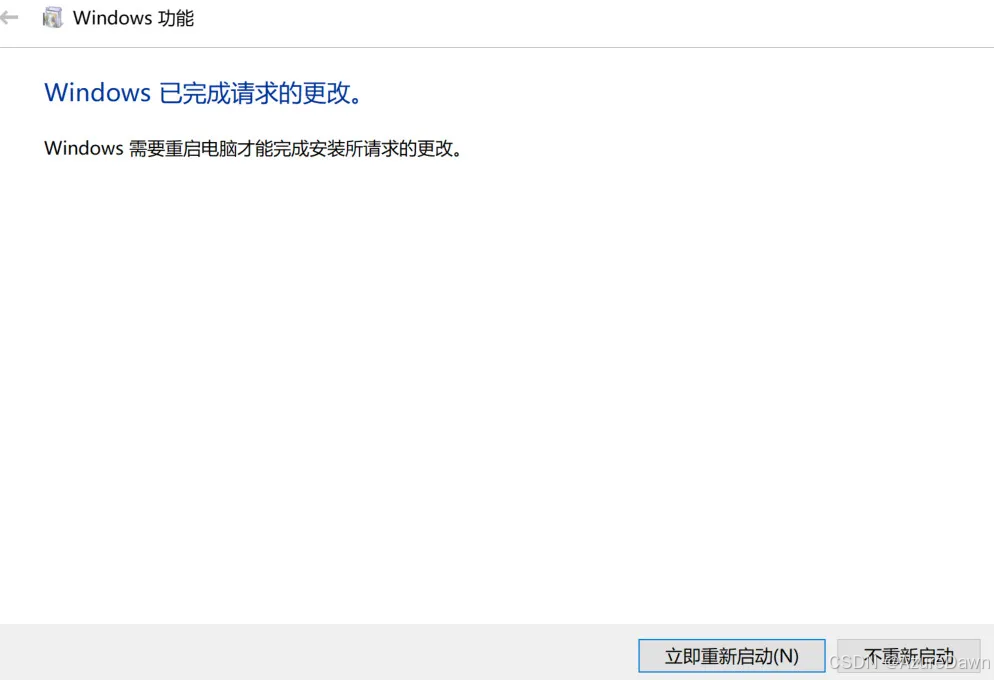
程序会自动安装相关底层软件,然后选择立即重新启动。
(4)下载Ubuntu
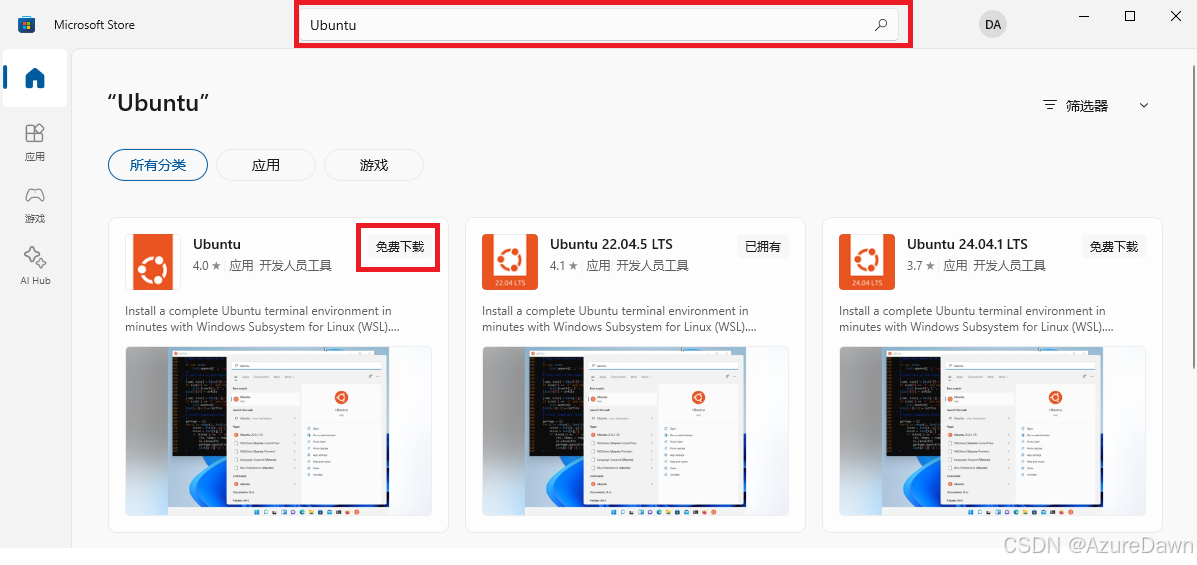
开始菜单旁“搜索”按钮,查找“app”并打开“Microsoft Store”,搜索“Ubuntu”,选择“Ubuntu”。点击免费下载并安装。
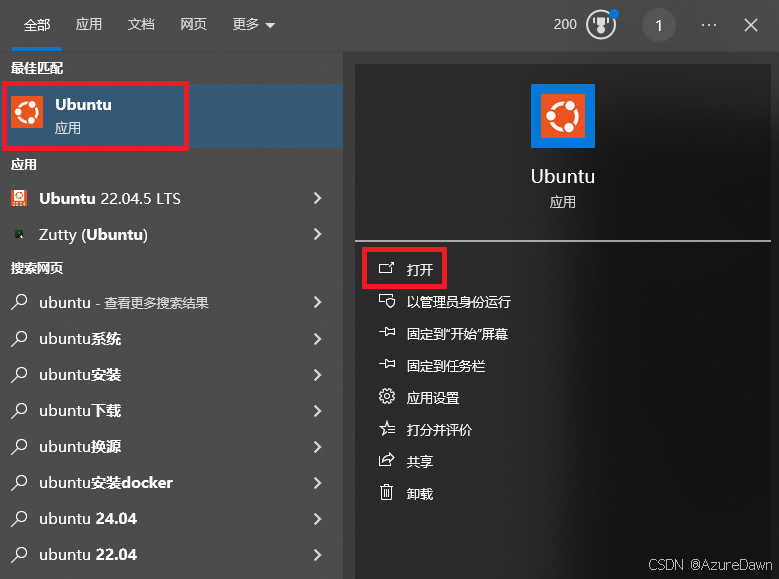
在开始菜单的搜索中输入ubuntu,即可打开Ubuntu。
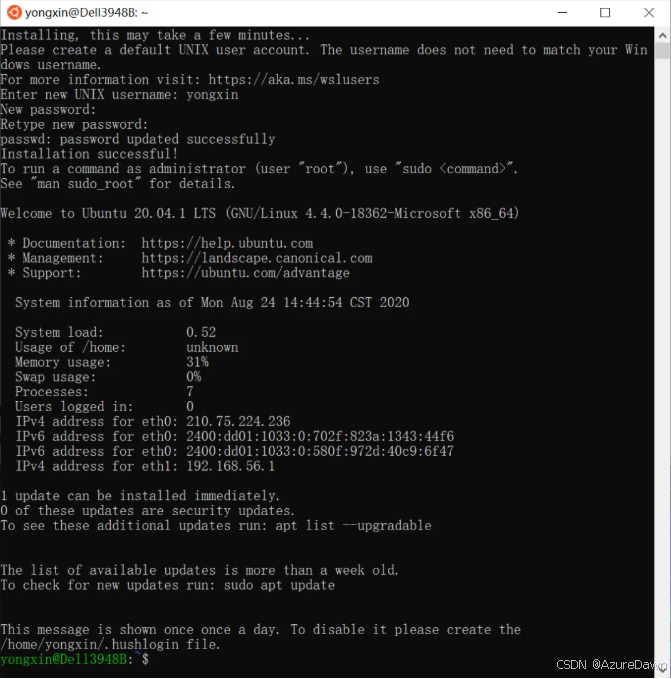
注:第一次启动会进行软件安装和布置,须等待几分钟。
提示输入用户名和密码。然后进入命令行模式,就可以开始下一步骤了。
2. Linux-conda环境安装
参考教程:【Conda】超详细的linux-conda环境安装教程_linux 安装conda-CSDN博客
超详细Ubuntu安装Anaconda步骤+Anconda常用命令_ubuntu 安装anaconda-CSDN博客
2.1 软件准备
(1)软件下载
下载网站:https://repo.anaconda.com/archive/index.html
根据自己的需要下载版本,我这里下载的是 Anaconda3-2024.10-1-Linux-x86_64.sh
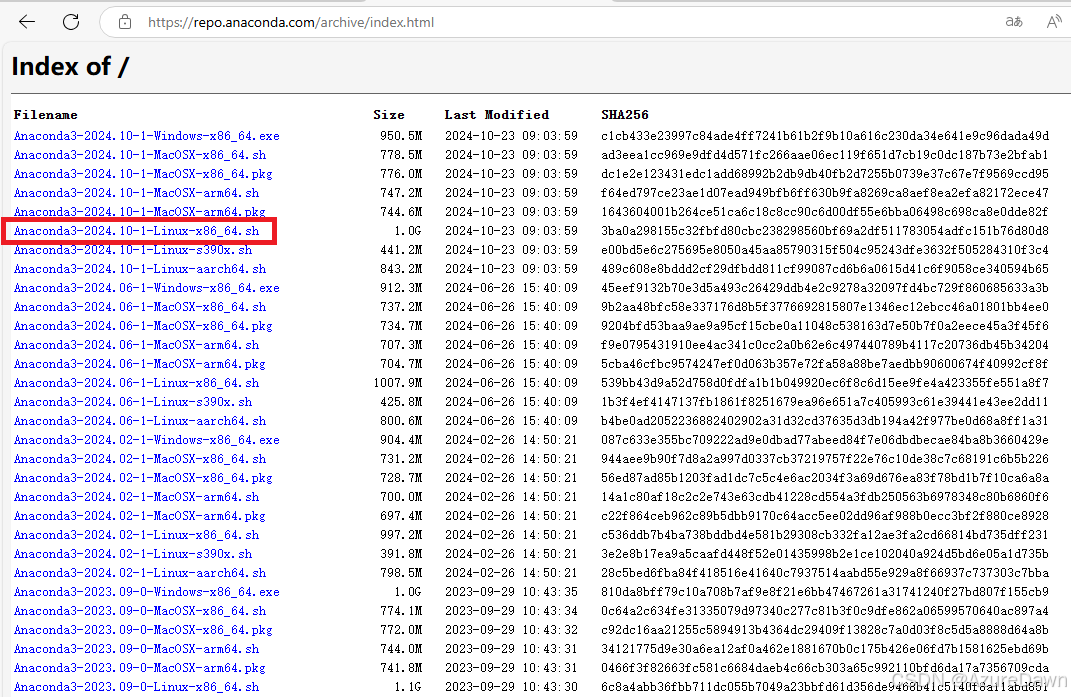
或者,可以复制下载的链接,直接在Ubuntu中下载:
wget -c https://repo.anaconda.com/archive/Anaconda3-2024.10-1-Linux-x86_64.sh(2)安装conda
在conda文件的目录下输入命令安装(将e/Anaconda3换为自己的下载地址)
cd /mnt/e/Anaconda3bash Anaconda3-2024.10-1-Linux-x86_64.sh一路回车,直到他要求输入yes。在输入yes后会提示安装位置,若选择默认可直接点击Enter键,conda便会开始安装。若想更换安装位置,可输入/mnt/e/Anaconda3进行更改(将e/Anaconda3换为自己的安装地址),确认点击Enter键。
(3)设置环境变量
编辑 ‘./bashrc’文件:
nano ~/.bashrc添加Anaconda路径到PATH环境变量,在文件末尾加上:
export PATH=/mnt/e/Anaconda3/bin:$PATH在nano中,ctrl+o保存文件,Enter确定后,ctrl+x关闭。刷新 ‘./bashrc’文件。
source ~/.bashrc最后验证conda是否生效:
conda --version2.2 镜像源设置
(1)conda配置
在Ubuntu中输入:
vim ~/.condarc点击s键进入vim的编辑。然后配置镜像,此处用的阿里云的镜像,可做参考。
channels: - https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/free/ - https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/main/ - https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/pytorch/ - https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/msys2/show_channel_urls: true ssl_verify: trueallow_conda_downgrades: true退出编辑可先点击Esc键进入命令模式,输入:wq并回车,即可实现保存并退出。
(2)pip配置
mkdir ~/.pipcd ~/.pip/vim pip.conf然后配置镜像:
[global] index-url = http://mirrors.aliyun.com/pypi/simple/ [install] trusted-host=mirrors.aliyun.com此时conda环境已经安装完成!
2.3 conda常用命令
# 创建虚拟环境conda create -n name python==3.8conda init bash # 激活环境conda activate name # 退出环境conda deactivate # 查看虚拟环境conda info --envs # 删除虚拟环境conda remove -n name --all # 删除所有的安装包及cache(索引缓存、锁定文件、未使用过的包和tar包)conda clean -y --all # 删除pip的缓存rm -rf ~/.cache/pip 二、测序数据的准备
测序数据的来源主要有两种,一种是从文章中获取编号,一种是直接使用公司返回的测序下机数据。
1. 从SRA数据库下载数据

2. 公司返回的测序下机数据
2.1 单端测序(Single-read)
数据格式应是:SRR******.fastq
2.2 双端测序(Paired-End)
数据格式应是:Sample1_R1.fastq.gz;Sample1_R2.fastq.gz; Sample2_R1.fastq.gz;Sample2_R2.fastq.gz…
三、对数据进行质控和过滤
进行质控和过滤的软件有很多,这里推荐使用国内OpenGene 最新开发的 fastp,速度非常快,且一步完成质控和过滤的流程,相当于之前经常用的fasqc质控+Trimmomatic过滤,可以自动检测adapter 序列,无需手动输入,非常方便。其质控和过滤效果也非常不错,可自行查阅相关资料。
fastp的运行模式分为单端测序和双端测序(当然目前主流都是双端),支持压缩文件(file.fq.gz)及fastq输入。
1. fastp安装
conda install -c bioconda fastp验证是否下载成功:
fastp --version2. 质控和过滤
进入工作目录(数据存放的地址):
cd /mnt/e/RNASeq-analysis/data/单端测序:
fastp -I SRR******.fastq -O SRR******_clean.fastq双端测序:
fastp -i Sample1_R1.fastq.gz -o Sample1_R1.clean.fastq.gz -I Sample1_R2.fastq.gz -O Sample1_R2.clean.fastq.gz当然,针对多个样本,我们也可以写个循环批量处理(所有文件须在同一个文件夹中):
for file in `ls *_R1.fastq.gz | perl -lpe \'s/_R1.fastq.gz//\'`; do fastp -i ${file}_R1.fastq.gz -I ${file}_R2.fastq.gz -o ${file}_R1.clean.fastq.gz -O ${file}_R2.clean.fastq.gz; done
四、参考基因组准备
参考教程:基因组注释文件(GFF,GTF)下载的五种方法_gtf文件下载-CSDN博客
1. 文件下载
参考基因组可以从NCBI、Ensemble、GENCODE、UCSC、iGenomes等网站下载。这里我选择从UCSC网站中获取。
下载网站:http://genome.ucsc.edu/cgi-bin/hgTables
设置参数如下,然后选择相应参数下载参考基因组及注释文件。
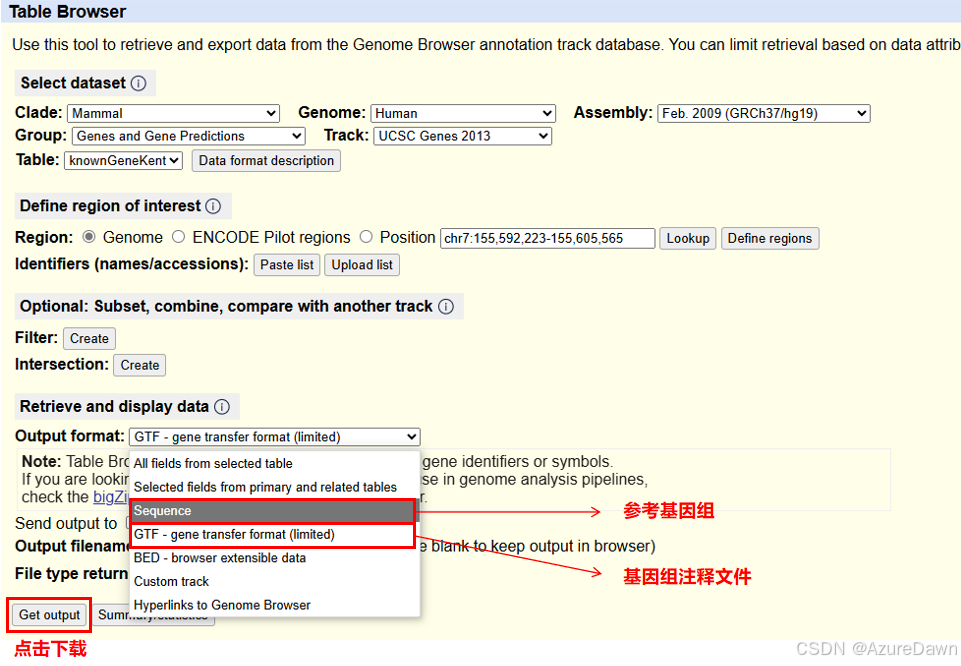
2. 文件准备
新建references文件夹:
mkdir references将参考文件置于references文件夹中:

五、比对到参考基因组
1. 构建索引
RNASeq reads 由于不包含内含子,所以来自外显子边界处的 reads被重新 mapping 回基因组时,会被中间的内含子分开,这种情况叫做 splice-alignment。比对的软件有很多,在这里推荐使用Hisat2或STAR。但STAR需要更大的内存,运行时间也更长,但准确性却相差不大。在这里使用Hisat2,使用conda安装Hisat2。
conda install hisat2进入工作目录:
cd /mnt/e/RNASeq-analysis/references/编写shell脚本:
vim hisat2-build.shvim编辑器的基础使用方法:
参考教程:Linux vi/vim | 菜鸟教程
进入编辑器后,点击键盘i键进入输入模式开始输入文本。输入完毕后点击Esc键退出输入模式,进入命令模式,再输入:wq指令(保存并退出),退出vim编辑器。
在vim编辑器中编写:
genome=/mnt/e/RNASeq-analysis/references/Species.genome.fastagenome_prefix=/mnt/e/RNASeq-analysis/references/Specieshisat2-build $genome $genome_prefix 1>/mnt/e/RNASeq-analysis/references/logs/hisat2-build.log 2>/mnt/e/RNASeq-analysis/references/logs/hisat2-build.err运行hisat2-build.sh:
nohup bash hisat2-build.sh &可以从logs/hisat2-build.log中查看进度。
2. 使用Hisat2进行比对
新建hisat2文件夹:
mkdir hisat2cd /mnt/e/RNASeq-analysis/hisat2编写shell脚本:
vim hisat2-run.sh在vim编辑器中编写:
genome_prefix=/mnt/e/RNASeq-analysis/Species# 写一个for循环进行批量运算# for paired -end, PE# 将以下目录替换成自己的文件目录for sample in $(ls /mnt/e/RNASeq-analysis/data/*_R1.clean.fastq.gz | perl -lpe \'s/\\/mnt\\/e\\/RNASeq-analysis\\/data\\///\' | perl -lpe \'s/_R1.clean.fastq.gz//\'); doif [ -f /mnt/e/RNASeq-analysis/data/${sample}_R2.clean.fastq.gz ]; thenhisat2 --new-summary -p 2 -x $genome_prefix -1 /mnt/e/RNASeq-analysis/89_FILE/${sample}_R1.clean.fastq.gz -2 /mnt/e/RNASeq-analysis/89_FILE/${sample}_R2.clean.fastq.gz -S /mnt/e/RNASeq-analysis/hisat2/${sample}.sam 2> /mnt/e/RNASeq-analysis/hisat2/${sample}.err;elseecho -e \"Warning: R2 file for $sample does not exists.\";fidone运行hisat2-run.sh:
nohup bash hisat2-run.sh &3. 比对结果压缩、排序及构建索引
新建sorted目录:
mkdir sortedcd /mnt/e/RNASeq-analysis/hisat2安装samtools:
sudo apt updatesudo apt install samtools -y验证是否安装成功:
samtools --version若正常显示版本信息,则安装成功;若报错 \"command not found\",说明未安装成功。
编写shell脚本:
vim sorted.sh在vim编辑器中编写:
for file in *.sam; dosamtools view -b $file > /mnt/e/RNASeq-analysis/sorted/${file%.sam}.bamsamtools sort /mnt/e/RNASeq-analysis/sorted/${file%.sam}.bam > /mnt/e/RNASeq-analysis/sorted/${file%.sam}.sorted.bamsamtools index /mnt/e/RNASeq-analysis/sorted/${file%.sam}.sorted.bamdone运行sorted.sh:
nohup bash sorted.sh &
六、计算表达量
1. 工具包下载
(1)安装R(若已有,可忽视;也可直接使用Rstudio):
sudo apt updatesudo apt install r-base(2)下载Biocmanager,安装Limma、Rsubread、edgeR及reshape2
在ubuntu中输入R并回车,进入R交互环境,运行代码。
# 安装 Bioconductor 管理器install.packages(\"BiocManager\")# 使用 Bioconductor 安装 limmaBiocManager::install(\"limma\")# 使用 Bioconductor 安装 RsubreadBiocManager::install(\"Rsubread\")# 使用 Bioconductor 安装 edgeBiocManager::install(\"edgeR\")# 安装 reshape2install.packages(\"reshape2\")(3)一一验证是否安装成功
library(BiocManager)library(limma)library(Rsubread)library(edgeR)library(reshape2)若均未显示报错,则证明安装成功。若下载失败,可检查是否为vpn问题,将系统代理关闭即可解决。
输入quit(),退出R环境。
2. 表达定量和标准化
使用R版本Subreads软件包中featureCounts计算表达量,其中Subread 的输入是 bam 文件和 gtf 文件,输出文件描述了落在每一个基因上的 reads 数目(read counts)。read counts还需要 TPM 和TMM标准化,这个标准化的步骤可以使用edgeR包里的fpkm函数。
新建featurecounts文件夹:
mkdir featurecountscd /mnt/e/RNASeq-analysis/featurecounts写一个循环运行featurecounts.R得到count文件:
for file in /mnt/e/RNASeq-analysis/sorted/*.sorted.bam; do Rscript featurecounts.R \"$file\" /mnt/e/RNASeq-analysis/references/genomic.gtf 10 \"$file\"; done3. 合并文件
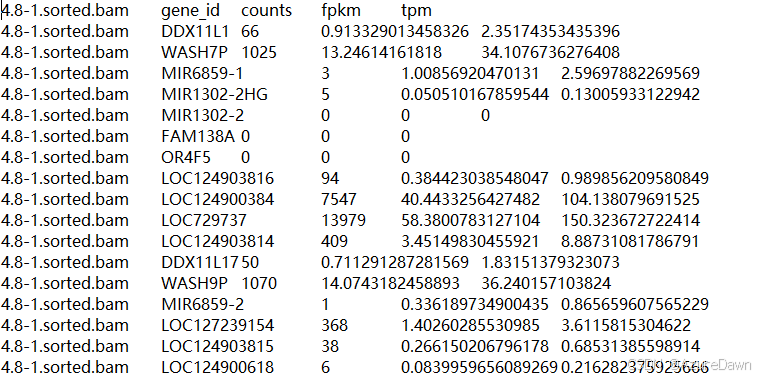
将所有count文件合并:
perl -lne \'if($ARGV=~/\\/mnt\\/e\\/RNASeq-analysis\\/sorted\\/(.*)\\.count/){print\"$1\\t$_\"}\' /mnt/e/RNASeq-analysis/sorted/*.count > merge.count第一列为样品名;第二列为基因名;第三列为counts数;第四列为fpkm值;第五列为tpm值
4. 生成矩阵
提取counts值及tpm值并生成矩阵:
awk -F\"\\t\" \'{print $1\"\\t\"$2\"\\t\"$3}\' merge.count > gene.countawk -F\"\\t\" \'{print $1\"\\t\"$2\"\\t\"$5}\' merge.count > gene.tpm编写R脚本:matrix_count.R
# 读取 count_matrix.txta=read.table(\'gene.count\',header=FALSE,sep=\"\\t\")# 赋予正确的列名colnames(a) = c(\'sample\', \'gene\', \'counts\')# 加载 reshape2 库library(reshape2)# 转换数据格式:gene_id 作为行,Sample 作为列counts = dcast(a, formula = gene ~ sample, value.var = \"counts\", fill = 0)# 保存到 gene_count.txtwrite.table(counts, file=\"gene_count.txt\", sep=\"\\t\", quote=FALSE, row.names=FALSE)运行R脚本:
Rscript matrix_count.R生成gene_count.txt。
第1列为基因名,第2列为Sample1对应的count数,第3列为Sample2对应的count数,以此类推。
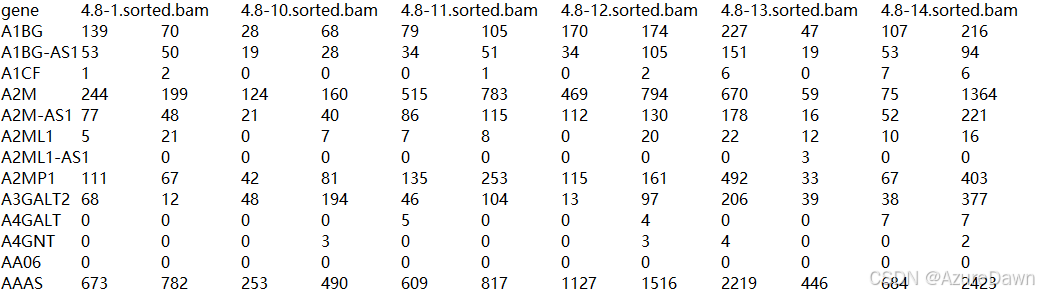
编写R脚本:matrix_tpm.R
a=read.csv(\'gene.tpm\',header=F,sep=\"\\t\")colnames(a)=c(\'sample\',\'gene\',\'tpm\')library(reshape2)counts=dcast(a,formula=gene~sample)write.table(counts,file=\"gene_tpm.txt\",sep=\"\\t\",quote=FALSE,row.names=FALSE)运行R脚本:
Rscript matrix_tpm.R生成gene_tpm.txt。
第1列为基因名,第2列为Sample1对应的tpm数,第3列为Sample2对应的tpm数,以此类推。
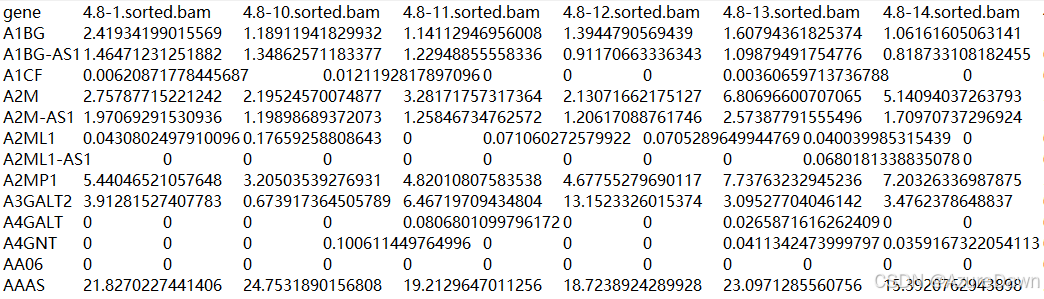
如有需要,可将文本文档中内容全选复制并粘贴至表格,即可得到gene_count.csv与gene_tpm.csv。
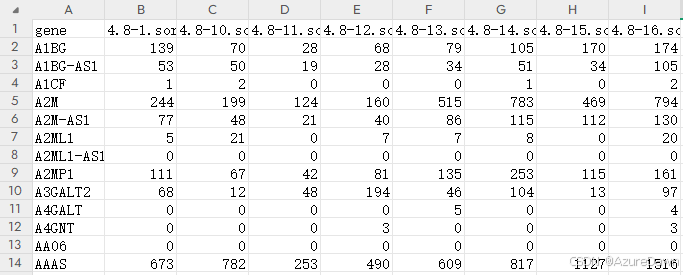
至此,基因表达矩阵提取完毕。