2025深圳杯&东三省数学建模 法医物证多人身份鉴定问题 保姆级教程讲解|模型讲解
2025深圳杯&东三省数学建模 法医物证多人身份鉴定问题 保姆级教程讲解|模型讲解
深圳杯&东三省联赛 D题保姆级教程思路分析
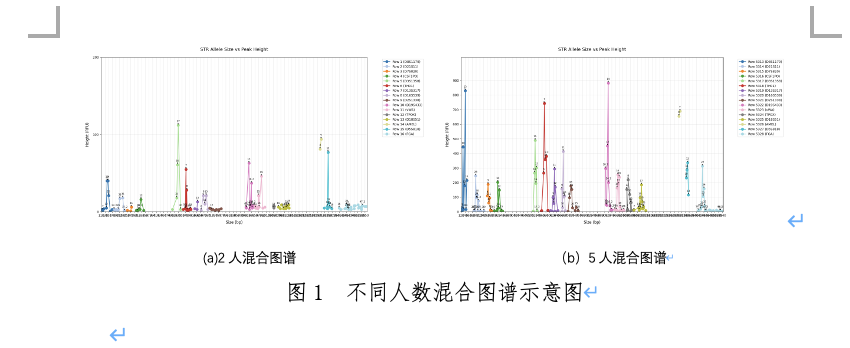
下面我将以背景介绍、数据集分析、问题分析的步骤来给大家讲解D题的具体思路。
1 背景介绍
法医物证鉴定是刑事侦查中确定犯罪现场遗留生物样本来源的核心技术,其中DNA分析技术(尤其是STR分析)因其高个体识别能力而被广泛应用。STR(短串联重复序列)通过检测基因座上等位基因的重复次数差异实现个体区分,每个个体在每个基因座上拥有两个等位基因(基因型)。混合STR图谱分析是多人案件中的关键挑战,需解决以下核心问题:
- 贡献者人数识别:混合样本中可能包含多名个体的DNA,错误估计人数会导致后续分析失效。
- 混合比例推断:不同贡献者的DNA比例影响等位基因峰高和重叠程度,需精准量化以区分基因型。
- 基因型反卷积:从混合信号中分离出各贡献者的基因型组合,需考虑等位基因共享和噪声干扰。
- 噪声抑制:实验噪声(如杂峰、基线漂移)会掩盖真实信号,需有效降噪以提高分析可靠性。
这里需要注意几点:1 数据复杂性:需处理多基因座、多峰值的混合信号,不同基因座的等位基因数量和峰高分布差异大。2 模型假设验证:如假定峰高与DNA量线性相关,需验证假设是否成立;需考虑低峰高等位基因的漏检风险。3 算法适应性:需设计能处理不同贡献者人数(如2-4人)、不同混合比例(如1:1、1:3)的通用方法。4 交叉验证机制:应结合附件3中已知基因型数据验证模型准确性,避免过拟合。5 生物学约束:基因型需符合孟德尔遗传规律(如每个基因座最多两个等位基因),需在模型中嵌入此类先验知识。6 噪声特性分析:需区分真实等位基因峰与实验噪声(如尖峰、宽峰),可能需结合信号平滑和阈值过滤。
2 数据集分析
以下为各附件核心字段说明及数据特性:
附件1/附件2/附件4(混合STR图谱数据)
- 字段:
- Sample File:样本编号
- Marker:基因座名称(如D8S1179)
- Dye:荧光染料类型(B/G等)
- Allele 1-Allele N:等位基因编号(整数或小数,OL表示Off-Ladder)
- Size 1-Size N:DNA片段长度(浮点数)
- Height 1-Height N:峰高值(整数,0表示无效峰)
- 数据特性:
- 每个样本在多个基因座上有多个等位基因峰
- 同一基因座存在多个等位基因(主峰+杂峰)
- Height=0或OL标记需过滤为噪声
附件3(贡献者基因型数据)
- 字段:
- Research ID:研究编号
- Sample ID:个体编号(1-50)
- 基因座列(如D8S1179):等位基因组合(如\"12,15\")
- 数据特性:
- 每个个体在每个基因座有且仅有两个等位基因
- 等位基因以逗号分隔(如\"29,31\")
- 用于问题3的基因型验证
可以这样做数据预处理:
1 混合图谱清洗:
将OL替换为NaN,过滤Height<=0的无效峰
按等位基因列拆分为长格式(每行一个等位基因)
保留数值型Size(排除非物理片段长度)
2 基因型数据转换:
拆分逗号分隔的等位基因(如\"12,15\"→Allele1=12, Allele2=15)
构建基因型集合(如{12,15})用于快速比对
下面是代码:
import pandas as pdimport numpy as np# --------------------------# 附件1/2/4预处理(混合图谱)# --------------------------def preprocess_mixture_data(filepath): # 读取原始数据 df = pd.read_excel(filepath, sheet_name=\"Sheet1\") # 清洗OL标记和Height=0的行 df = df.replace(\"OL\", np.nan) for col in df.filter(like=\'Height\').columns: df[col] = pd.to_numeric(df[col], errors=\'coerce\') df.loc[df[col] <= 0, col] = np.nan # 转换宽表为长表(行:样本-基因座-等位基因) allele_cols = [c for c in df.columns if \'Allele\' in c] melted_dfs = [] for idx in range(1, len(allele_cols)+1): temp_df = df[[\'Sample File\', \'Marker\', \'Dye\',f\'Allele {idx}\', f\'Size {idx}\', f\'Height {idx}\']] temp_df.columns = [\'Sample\', \'Marker\', \'Dye\', \'Allele\', \'Size\', \'Height\'] melted_dfs.append(temp_df.dropna(subset=[\'Height\'])) combined_df = pd.concat(melted_dfs).reset_index(drop=True) # 过滤有效等位基因(Size为数值) combined_df[\'Size\'] = pd.to_numeric(combined_df[\'Size\'], errors=\'coerce\') combined_df = combined_df.dropna(subset=[\'Size\']) return combined_df# --------------------------# 附件3预处理(已知基因型)# --------------------------def preprocess_genotype_data(filepath): df = pd.read_excel(filepath, sheet_name=\"Sheet1\") # 拆分等位基因列为两列 genotype_dfs = [] for marker in df.columns[2:]: temp_df = df[[\'Research ID\', \'Sample ID\', marker]].copy() temp_df[[\'Allele1\', \'Allele2\']] = temp_df[marker].str.split(\',\', expand=True) temp_df[\'Marker\'] = marker genotype_dfs.append(temp_df[[\'Sample ID\', \'Marker\', \'Allele1\', \'Allele2\']]) combined_df = pd.concat(genotype_dfs).reset_index(drop=True) # 转换为集合格式便于匹配 combined_df[\'Genotype\'] = combined_df.apply( lambda x: {x[\'Allele1\'], x[\'Allele2\']}, axis=1 ) return combined_df[[\'Sample ID\', \'Marker\', \'Genotype\']]# 示例调用mixture_df = preprocess_mixture_data(\"附件1.xlsx\")genotype_df = preprocess_genotype_data(\"附件3.xlsx\")数据处理可以用的可视化代码有:
等位基因峰高分布 - 箱线图
可以观察不同基因座的峰高分布差异,识别离群峰(可能为噪声或低贡献者信号)
import seaborn as snsimport matplotlib.pyplot as plt# 数据准备(假设已预处理为mixture_df)plt.figure(figsize=(12, 6))sns.boxplot(data=mixture_df, x=\'Marker\', y=\'Height\', palette=\'viridis\')plt.xticks(rotation=45)plt.title(\"Peak Height Distribution by Marker\")plt.show()等位基因数量统计 - 堆积柱状图
用途:统计每个样本/基因座的等位基因数量,辅助判断贡献者人数(如每个基因座最多2N个等位基因,N为人数)
# 统计每个基因座的等位基因数量allele_counts = mixture_df.groupby([\'Sample\', \'Marker\'])[\'Allele\'].ncount().unstack()plt.figure(figsize=(14, 6))allele_counts.plot(kind=\'bar\', stacked=True, colormap=\'tab20\')plt.title(\"Allele Count per Marker and Sample\")plt.xlabel(\"Sample\")plt.ylabel(\"Number of Alleles\")plt.legend(title=\'Marker\', bbox_to_anchor=(1.05, 1))plt.show()3 问题分析
问题一分析与求解:
建模思路:
贡献者人数识别需基于以下生物学约束与数据特性:
- 等位基因数量限制:每个贡献者在同一基因座贡献至多2个等位基因,N人混合样本中每个基因座的等位基因数≤2N。
- 峰高比例性:等位基因峰高近似与贡献者DNA量(即混合比例)成正比。
- 噪声干扰:存在低峰高杂峰(Height接近0)需过滤。
建模流程:
步骤1:数据预处理
- 输入:附件1原始数据(多基因座混合STR图谱)
- 操作:
- 过滤无效峰(Height≤阈值如50 RFU)
- 合并同一基因座的等位基因列表
- 输出:结构化数据格式(样本×基因座×有效等位基因列表)
步骤2:特征提取
- 关键特征:
- 每个基因座的等位基因数(num_alleles)
- 峰高变异系数(height_cv)
- 峰高熵(height_entropy)
步骤3:人数估计模型
模型1:最大等位基因数法(Baseline)
- 原理:取所有基因座中等位基因数最大值,计算N = ceil(max(num_alleles)/2)
模型2:高斯混合模型(GMM)
- 原理:假设不同人数对应的特征(如num_alleles, height_cv)服从高斯分布,使用EM算法拟合混合分布。
步骤4:模型集成与投票
- 策略:对多个基因座结果取众数(如多数基因座支持2人则最终输出2)
改进方向可以是:

上面用到的模型公式有:

后续思路、代码等持续更新。
其中更详细的思路、各题目思路、代码、讲解视频、成品论文及其他相关内容,可以点击下方卡片哦:


