论文笔记八【3D equivariant diffusion for target-aware molecule generation and affinity prediction】
论文笔记_大作业PPT: 这是我自己做的配套PPT,仅供参考 (gitee.com)
今天给大家介绍的论文是《3D equivariant diffusion for target-aware molecule generation and affinity prediction》,发表在2023年的ICLR会议中,据文章所述,这是首个用于靶点感知药物设计的概率扩散模型。
一、背景
现有主流的分子生成模型通常基于SMILES字符串(一维)或图(二维),但这两种表示方式都未考虑分子三维空间中的相互作用。随着结构生物学和蛋白质结构预测的最新进展,越来越多的结构数据得以获取,为机器学习算法在三维结合复合物中直接设计药物提供了新的机会——
如将三维空间表示为体素化网格,并使用三维卷积神经网络(3D CNN)对蛋白和分子进行建模。然而,该方法不具备旋转等变性,无法充分捕捉三维归纳偏差。此外,体素化操作的计算量会随着口袋大小的立方增长,导致较差的可扩展性。
一些先进方法通过不同的建模技术实现 SE(3)-等变性(Special Euclidean group in 3D)。然而,这些方法采用自回归采样策略,即基于学习到的原子类型和原子坐标概率密度逐个生成原子。这类方法存在多个局限性,举其中一个例子——模型在采样时设定了一种不自然的生成顺序,无法考虑整个三维结构的整体概率。例如,在已将 n−1 个碳原子放置在同一平面的情况下,模型较容易正确放置第 n 个原子以形成苯环。但对于前几个原子的放置,由于缺乏足够的上下文信息,模型难以做出准确预测,容易生成不合理的结构片段。
因此,作者提出了 TargetDiff,这是一种端到端的三维全原子扩散模型,以非自回归方式生成靶点感知分子。具体而言,作者将蛋白结合口袋和小分子表示为三维空间中的原子点集,每个原子都关联一个三维笛卡尔坐标。作者为连续的原子坐标和离散的原子类型定义了一个扩散过程,其中噪声逐步加入,并利用 SE(3)-等变图神经网络学习联合生成过程,该网络交替更新分子的原子隐藏嵌入和原子坐标。
二、模型框架
因为论文中并没有画模型图,只给出了算法,所以这部分结合算法来讲。
2.1算法1:该扩散模型的训练过程
输入蛋白-配体绑定数据集。在这里,P 代表蛋白质的结构信息,M 代表配体的分子信息。模型的最终目标是训练一个神经网络θ,它可以通过输入的信息生成相应的分子。首先要从均匀分布(0,T)中采样扩散时间t,t=0 代表初始状态(无噪声),t=T 代表完全扩散(最大噪声),这样可以让模型在训练时学习如何处理不同程度的噪声。然后将蛋白质分子的质心(Center of Mass, CoM)对齐到原点,确保训练过程中蛋白质的坐标不会因为位移而影响预测结果。
之后进行原子坐标的扩散,不断扰动原子坐标,逐步向高斯噪声转变,模拟一个从真实分子结构到噪声分子的过程,然后训练模型学会反向去噪。x0是初始(无噪声)的分子坐标。xt是扩散到时间 t 后的分子坐标。at是时间步 t 处的扩散系数(控制噪声强度,其值越小,扩散扰动越明显),ϵ∼N(0,I) 是从标准正态分布采样的噪声。
然后进行原子类型的扩散,不断扰动原子类型,逐步将原子类型向均匀分布转换,使得原子类型信息在扩散过程中变得模糊,然后让模型学会如何从噪声中恢复它们。at控制扰动强度,K代表有K种原子类型。
现在通过经网络θ接收当前噪声状态[xt,vt],预测去噪后的原始分子及其类型[x0,v0]。再就是计算后验概率、MSE损失和KL散度损失,然后根据梯度下降法更新神经网络参数,如果没有收敛就继续训练。

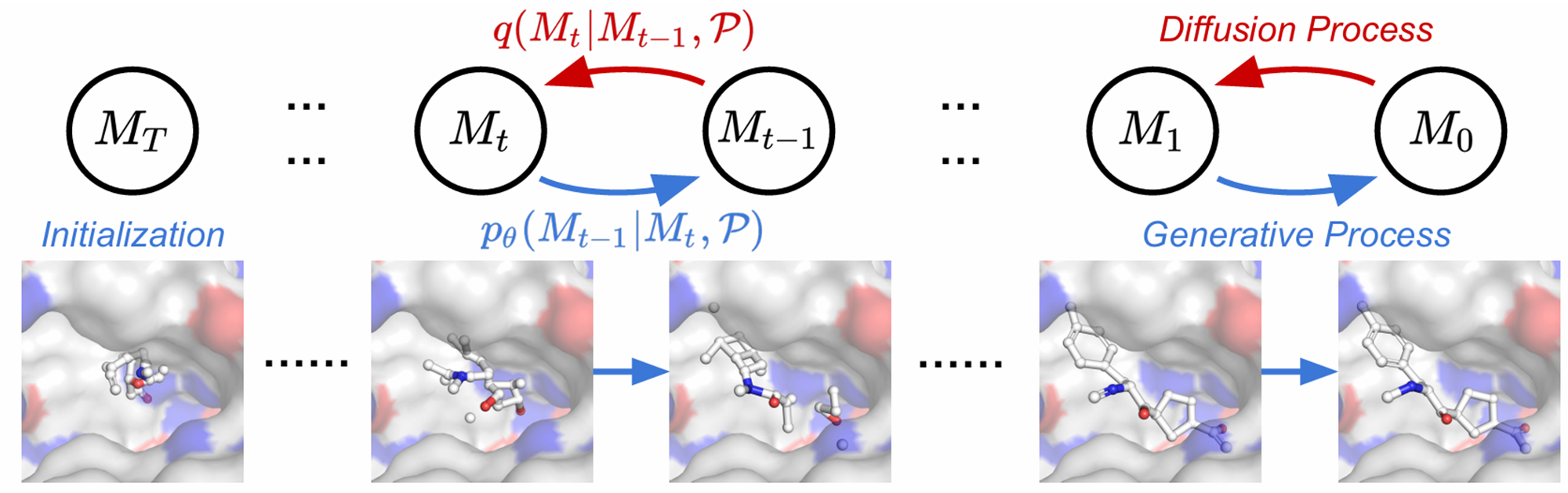
2.2算法2:使用扩散模型生成新的配体分子
为什么叫采样而非分子生成:采样(sampling)本质上是从一个概率分布中抽样生成具体实例的行为。在分子生成任务中,作者的目标是学习一个模型,使其能够生成类似真实分子的结构。这意味着作者希望模型学会一个分布p(x),其中,x 表示分子结构。这个分布通常很复杂,不能直接写出显式公式。去噪过程就对应着在每一步的后验分布p(xt−1∣xt) 中采样(sampling)一个状态。因为每一步都是从一个分布中抽样而非确定选择,即便输入同一个蛋白质口袋,不同采样轨迹可能生成不同的配体,从而保证多样性。这与“直接预测”不同,采样过程可以自然地探索多个可能的解。所以一般都使用“采样”来描述分子生成,即模型不是直接输出某个确定的分子结构,而是通过从一个学到的概率分布中抽样得到一个分子结构。

输入蛋白质结合位点P和训练好的神经网络θ,输出配体分子结构M。首先根据蛋白结合口袋的大小,从先验分布(prior distribution)中采样生成的配体原子数。比如较大的结合口袋可能生成更大的配体,较小的结合口袋可能生成较小的配体,之后同样的将蛋白质分子的质心(Center of Mass, CoM)对齐到原点,确保训练过程中蛋白质的坐标不会因为位移而影响预测结果。
然后采样原子的初始噪声状态,从标准正态分布N(0,I)中采样表示分子的坐标初始状态是纯噪声,Gumbel-Softmax 采样说明最初的原子类型是从均匀随机分布中抽取的,这个步骤就是在完全噪声的空间里随机初始化一个分子。
进行逐步去噪,从T开始,直到t=1,从纯噪声状态逐渐生成合理的分子结构——其中,每一次去噪都会预测一次当前状态下的“无噪声”分子x0,同时从后验分布pθ(xt-1|xt,x0)采样新的坐标xt-1,从后验分布pθ(vt-1|vt,v0)中采样新的原子类型vt-1,通过这样的迭代逐步修正原子坐标和原子类型,直到收敛至合理的分子坐标和合理的化学元素。
2.3具体参数
TargetDiff包含 9 层等变(equivariant)神经网络,每层是一个 Transformer,隐藏维度为 128,注意力头数为 16。设置扩散步数为 1000,模型使用 Adam 优化器进行训练,初始学习率为 0.001,batch_size为 4,作者对原子类型的损失乘以一个因子 α = 100,以平衡坐标损失和类型损失的尺度。在训练过程中,作者对蛋白质原子坐标添加标准差为 0.1 的高斯噪声作为数据增强。学习率采用指数衰减策略,衰减因子为 0.6,最低学习率为 1e-6。当验证损失在连续 10 次评估中没有提升时触发衰减。每 2000 步训练会进行一次评估。作者在一张 NVIDIA GeForce GTX 3090 GPU 上训练模型,训练可在 24 小时内、20 万步内收敛。
三、实验结果
首先,作者绘制了所有原子间距离及碳-碳键距离的经验分布(见图),并将其与参考分子的经验分布进行对比。从整体原子间距离来看,TargetDiff 能很好地捕捉整体分布,而 AR 和 Pocket2Mol 在小原子间距上存在过度表现(over-representation)。由于 liGAN 受到其有限的体素化分辨率限制,它只能捕捉整体形状,而无法准确建模具体的模态(modes)。
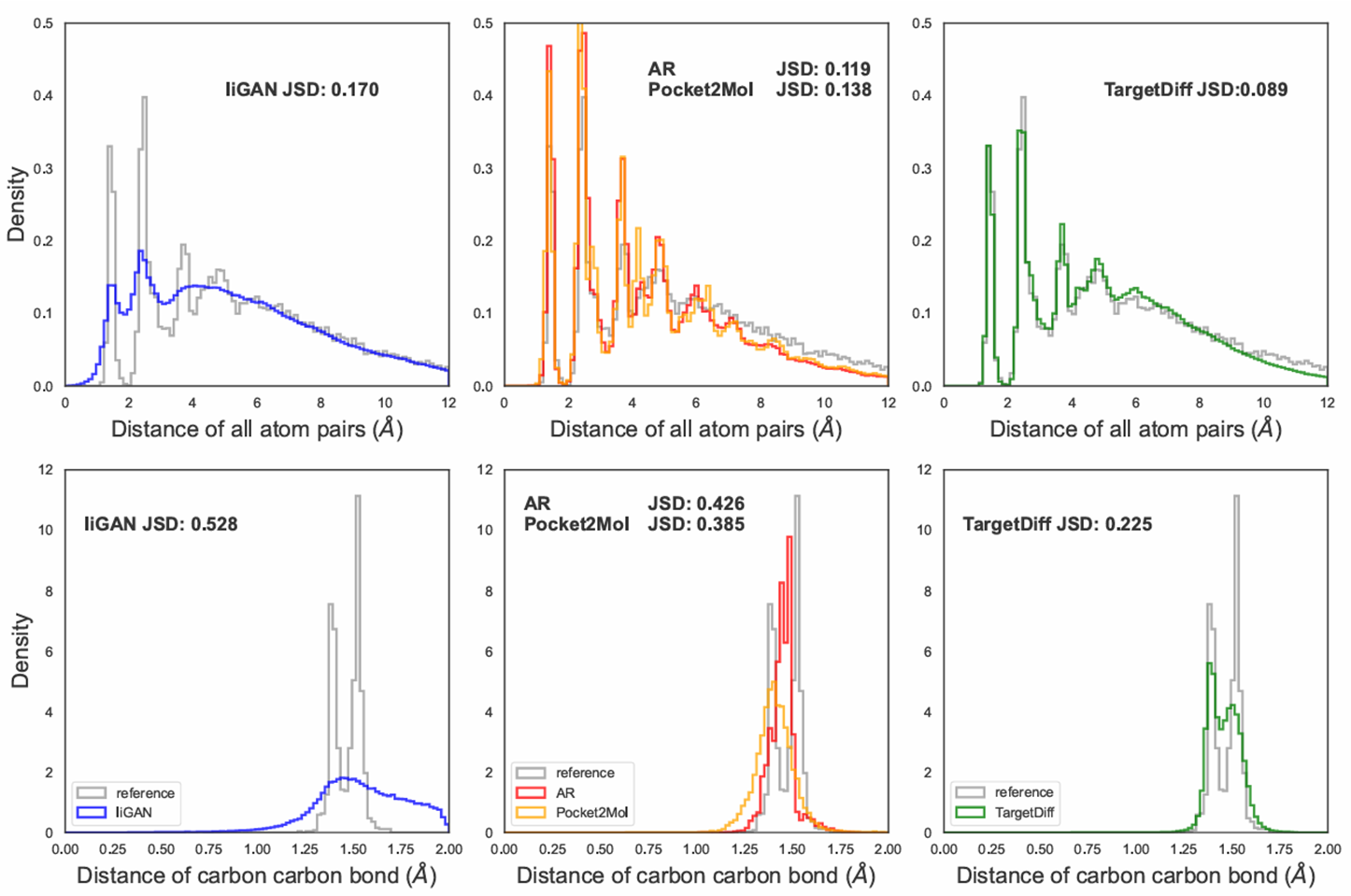
类似地,在参考分子的结构中,不同类型的碳-碳键形成了两种代表性距离模式。在 TargetDiff 生成的分子结构中,作者仍能观察到这两种模式,而在 liGAN、AR 和 Pocket2Mol 生成的分子中仅能观察到单一模式。作者进一步通过 Jensen-Shannon 散度(JSD)测量不同模型生成的分子结构与参考分子键长分布的相似度。结果表明,TargetDiff 在所有主要键类型上均明显优于其他方法。
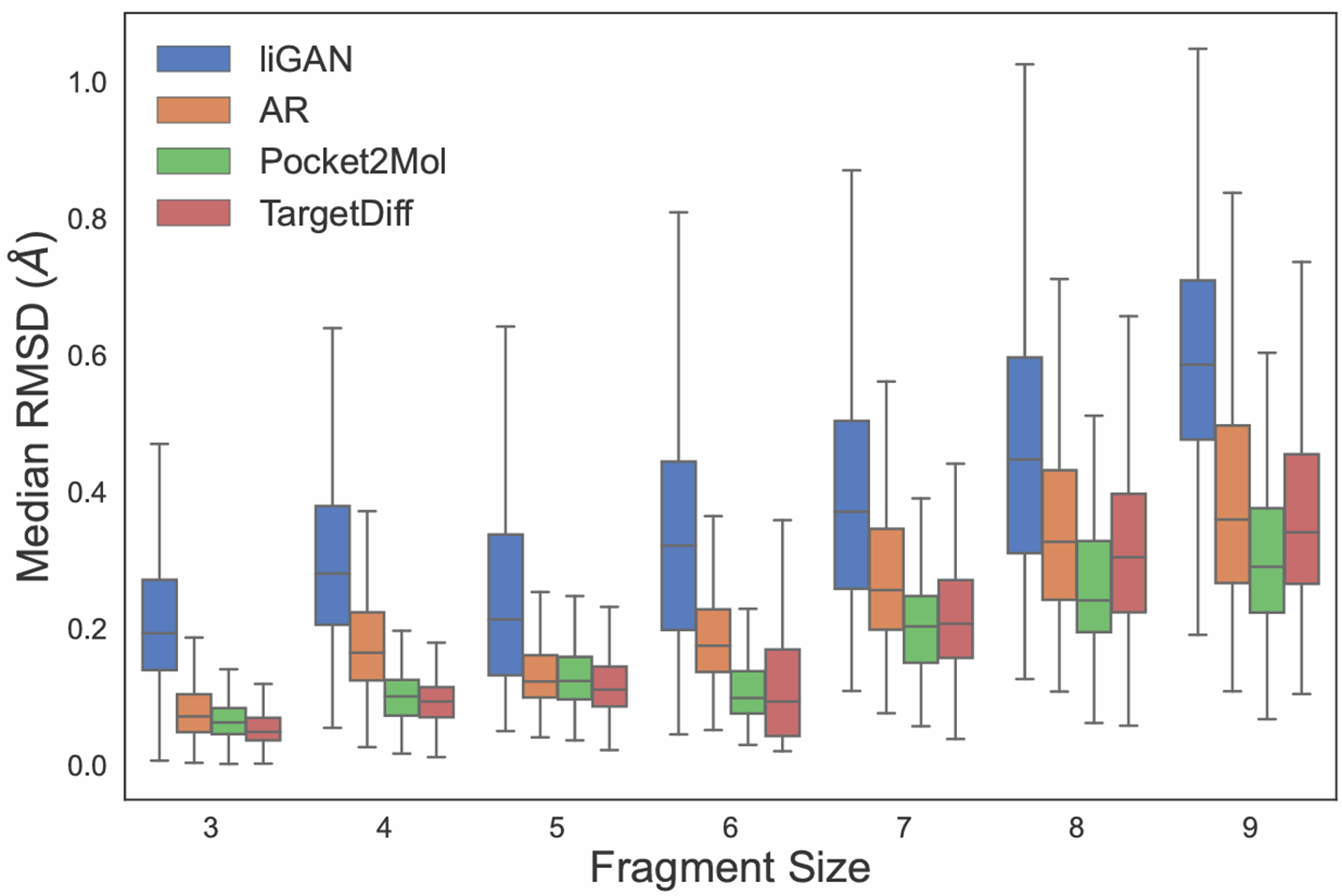
其次,作者探讨了 TargetDiff 是否能够一致性地生成刚性子结构(rigid sub-structure)或片段(fragment),例如所有碳原子在苯环中是否共面。为了评估这种一致性,作者使用 Merck 分子力场(MMFF)(Halgren, 1996)优化生成的分子结构,并计算 MMFF 优化前后刚性片段(不含可旋转键)的均方根偏差(RMSD)。如图所示,TargetDiff 能够生成更加一致的刚性片段。
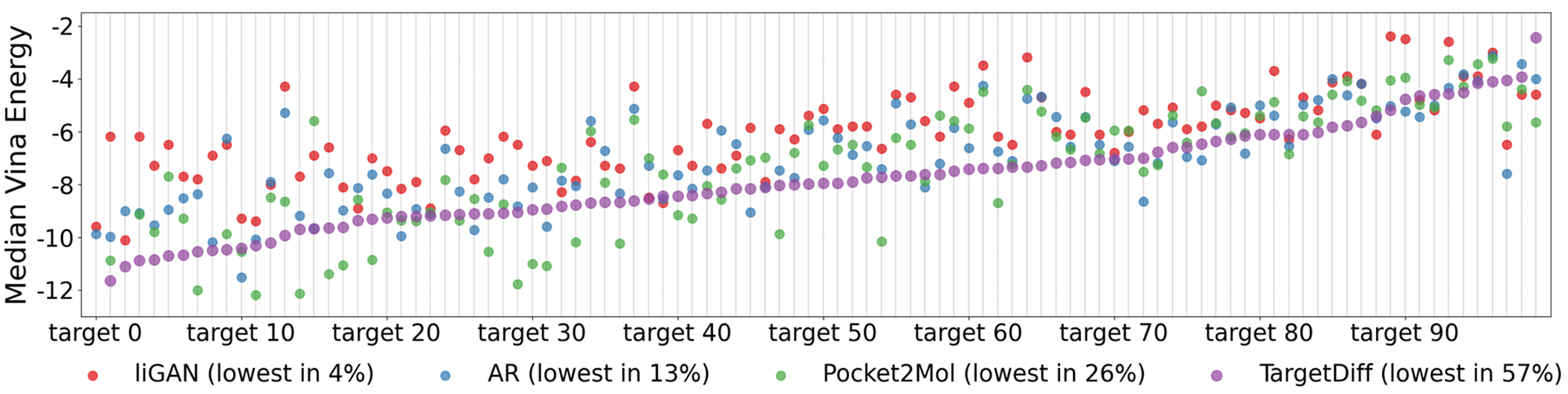
图显示了所有生成分子在100个测试结合口袋(binding pocket)中的中位 Vina 能量(Vina energy),该值由 AutoDock Vina (Eberhardt et al., 2021) 计算。基于 Vina 能量,TargetDiff 生成的分子在 57% 的目标上表现出最佳的结合亲和力,而 liGAN、AR 和 Pocket2Mol 仅在 4%、13% 和 26% 的目标上表现最佳。
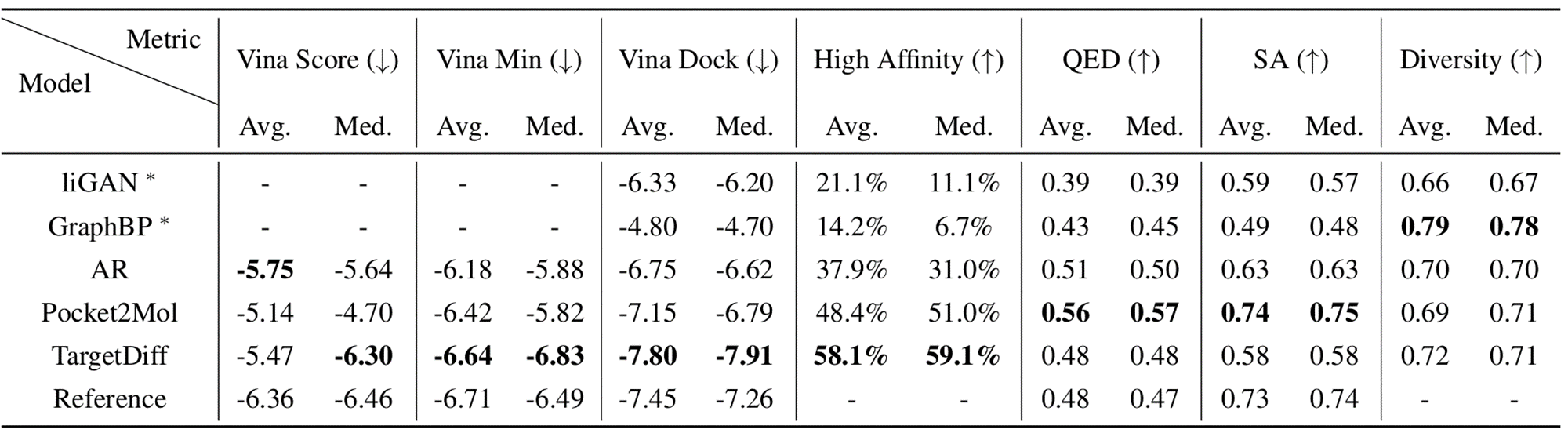
在高亲和力配体(high-affinity binder)方面,平均 58.1% 的 TargetDiff 生成分子比参考分子具有更好的结合亲和力,这一比例明显优于其他基线方法(见表)。此外,作者在表中计算了 Vina Score 和 Vina Min,其中 Vina Score 直接计算或在不重新对接(re-docking)的情况下进行局部优化。这些指标直接反映了模型生成的 3D 分子质量,TargetDiff 也在这些指标上优于所有基线方法。
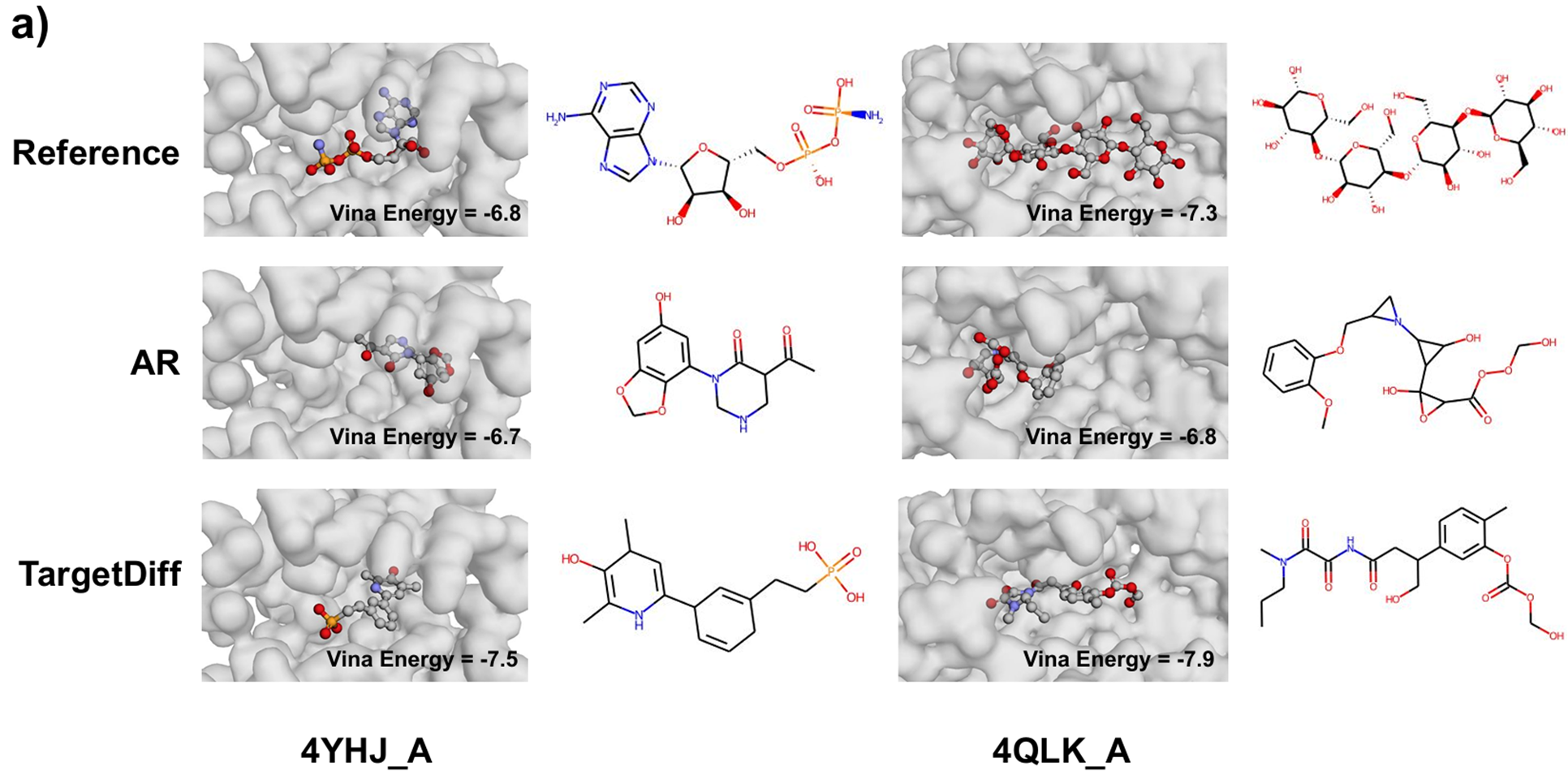
为了更深入理解各模型生成分子的差异,作者从每个模型生成的分子中选取样本,并比较其在两个结合口袋中的表现,TargetDiff 在这两个口袋上的表现均优于 AR。如图所示,TargetDiff 生成的分子能够填满整个结合口袋,而 AR 生成的分子只能部分覆盖口袋空间,可能因此降低了对目标的特异性,导致“非靶标效应”(off-target effects)。以 AR 生成的 4QLK A 口袋分子为例,尽管其原子数量与 TargetDiff 生成的分子相近(27 vs. 29),但 AR 的前沿网络(frontier network)在放置原子时过度深入结合口袋,而未考虑整体结构,从而导致无法完全覆盖结合口袋,最终导致较差的结合亲和力。
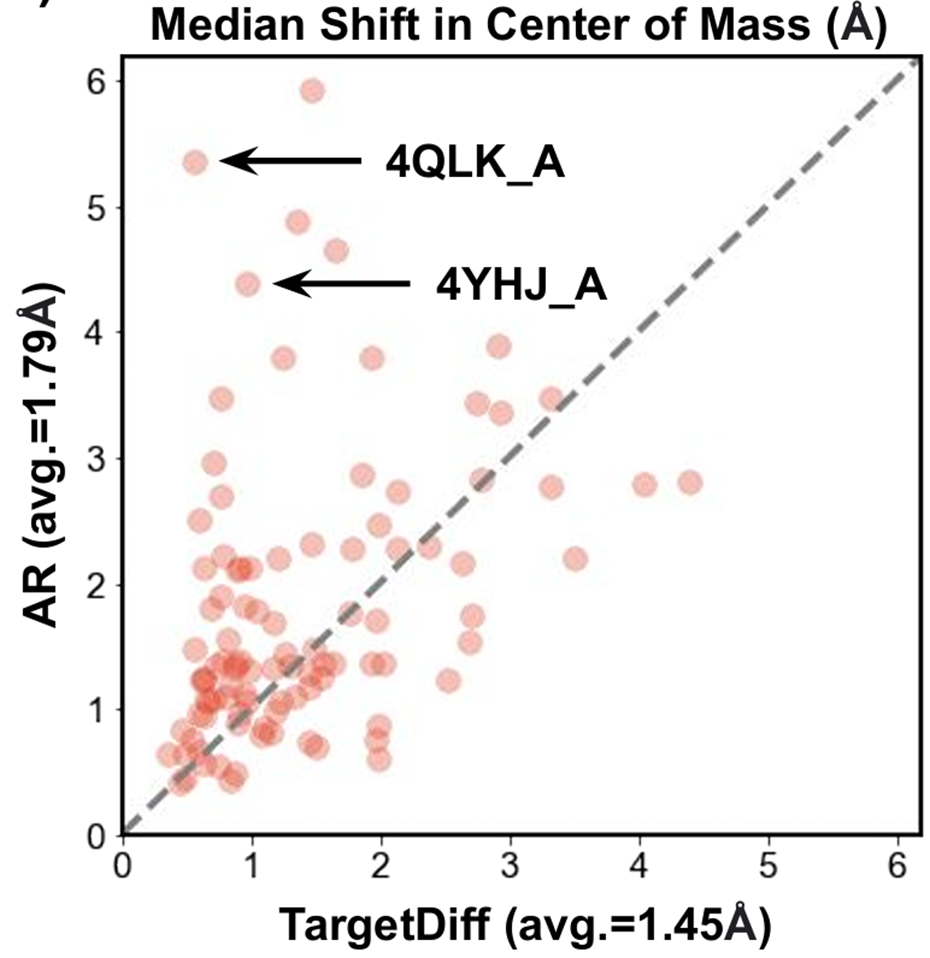
为了进一步量化这种影响,作者测量了参考分子的质心(CoM)与生成分子的质心之间的距离。如图所示,由于 AR 采用了顺序生成的方式,其 CoM 偏移量更大(1.79 Å vs. 1.45 Å),导致较差的结合构象(binding pose)和较低的结合亲和力。
四、总结与思考
针对扩散模型,对于幻觉问题、大量的三角形和苯环的生成(如文中的七元环),这样奇怪的拓扑结构还需要进一步限制。本文只有针对原子的扩散,如果将键生成纳入扩散过程会是一个有趣的方向,这样作者就可以跳过键推断算法,而且更有整体一致性。
对于写文章来说,这篇文章也就是把扩散模型应用到分子生成任务中,然后稍微加一点自己的工作,客观上不算难度大、工作量大,但是却能投到顶会上。所以我感觉科研要紧跟热点,尝试用最新的方法放到自己的领域,如果效果不错,就能试着发文章了。


